E. K. Davies and C. J. Davies
Treweren Consultants Ltd, Holmleigh, Evesham Road, Harvington, Evesham WR11 8LU, UK
Structure-based virtual screening has a reputation for generating a significant percentage of false positives. Traditionally developers have placed an emphasis on reproducing crystallographic small molecule-protein complexes [1] and used enrichment measures over random screening as a success measure [2]. Our interest in false positives arose out of analysing the result from the CAN-DDO and Find-a-Drug [3,4] projects which screened billions of molecules using the THINK software which we developed. The implementation of the docking algorithms used in the THINK software are enhanced and more efficient than the pharmacophore based method which was implemented in the Chem-X software [5].
There is general acceptance that proteins are flexible: unfolding and refolding as well as exhibiting smaller movements in side-chains, loops etc. Nonetheless, most docking software assumes that the conformation observed in the crystal structure is biologically relevant although some users do consider a few conformational variants. The term induced fit is sometimes used to describe docking where there are significant changes in the protein conformation, for instance to accommodate a larger molecule in a P450 site. In this paper we describe new methodology which is consistent with larger and more general protein conformational changes than previously expected.
The pharmacophore profiling technology implemented in Chem-X has proven to be effective [6]. Developments in computer processor speeds and available memory have allowed better algorithms to be used for the implementation in the THINK software. More classes of interaction centre (up to 12) and more graduations of distances or bins (up to 31) are used when constructing a profile, as listed in Table 1. In addition, rather than use a single bit to indicate the presence or absence of a pharmacophore in the profile a counter or population is used. This overcomes a comparison problem between pharmacophores which are similar but would otherwise set different bits because the distances fall either side of a bin boundary.
| Interaction Centre Types | Distance bin graduations (Å) |
| H-bond donor | <= 3.0 |
| H-bond acceptor | <= 4.0 |
| Acid | <= 5.0 |
| Base | <= 6.0 |
| Positive charge | <= 7.0 |
| Negative charge | <= 9.0 |
| Aromatic ring | <= 11.0 |
| Lipophilic group | <= 14.0 |
| Lewis Acid | <= 17.0 |
| Lewis Base | <= 20.0 |
| > 20.0 |
Table 1: Default pharmacophore interaction types and distance bin graduations
When a distance is within a tolerance of a bin boundary, both bins are populated in proportion to the distance from that boundary. Furthermore, the population is adjusted based on the Boltzman population of the conformation exhibiting the pharmacophore which has the consequence that pharmacophores which are exhibited by high energy conformations are eliminated and the profile is dominated by data from low energy conformers.
Given a set of molecules which bind competitively at the same receptor site one might expect similar pharmacophores to be exhibited. Furthermore, one would expect a pharmacophore profile to identify such pharmacophores which in turn can be used to search or mine combinatorial chemistry libraries, corporate and catalogue databases for molecules to test. We observed that such searches gave large numbers of false positives and sought a means of adding a volume constraint to the search.
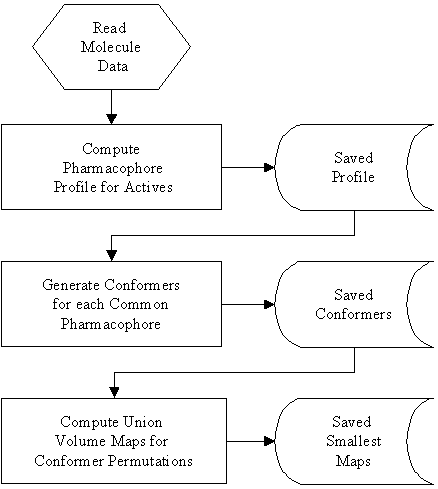
The Active Volume Constraint implementation in the THINK software generates a union volume for a conformer of each active molecule which exhibits a pharmacophore, using the procedure outlined in figure 1.
Figure 1: Procedure for computation Active Volume Constraint Maps.
Flexible molecules often have several conformations exhibiting the same pharmacophore which may differ by the orientation of another part of the molecule. Consequently, it is not unusual to have large numbers of permutations of conformers, one for each molecule, for which union volume maps are constructed. Fortunately, the smallest, most constraining volume maps are of interest and it is not necessary to store all the volume maps. Figure 2 shows an example of such a volume constraint map with one of the molecules which defined it and part of the pharmacophore. The implementation is fully automated and uses temporary files to store the large amount of intermediate data.
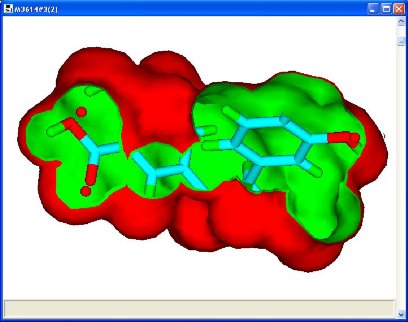
Figure 2: Volume constraint map with one of the molecules used to define it and a pharmacophore (3 red spheres)
Table 2 details 7 protein targets which were included in the Find-a-Drug project predicting a number of virtual hits that have been tested under the National Cancer Institute [7] cell-line assays. Those with activities below �M were considered active.
Active volume constraint maps were calculated for those pharmacophores which were present in at least 90% of the molecules. The volume constraints and the pharmacophores were then used to search the virtual hits. The results are shown in figure 3. In all cases the actives were retrieved and 318 of the 330 false positives (96.4%) were eliminated. This increased the true positive hit rate to 83.8%.
| PDB ID | Protein | Hits | Actives |
| 1B8Y | Stromelysin (MMP-3) | 48 | 6 |
| 1NVS | Checkpoint Kinase (Chk1) | 74 | 13 |
| 1Q4P | C-SRC tyrosine kinase (SH2 domain) | 82 | 14 |
| 1QZY | Methionine aminopeptidase 2 | 81 | 15 |
| 1UU3 | 3-phosphoinositide dependent protein kinase-1 | 35 | 5 |
| 1V8K | Kinesin-like protein KIF2C | 45 | 5 |
| 1FGI | Fibroblast growth factor receptor | 2 | 4 |
Table 2: Protein targets, virtual hits and actives (true positives).
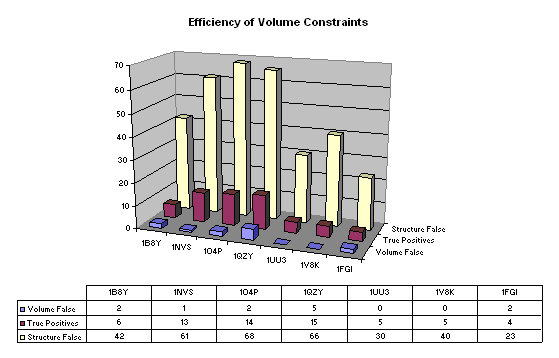
Figure 3: Pharmacophore with volume constraint searches. 'Volume False' are the false positive hits accepted by the active volume constraint and 'Structure False' are the false positive hits from structure based virtual screening included for comparison.
In order to ascertain whether these results were spurious we scrambled the activity data (maintaining the proportion of actives/inactives) and repeated the calculations. The results are shown in figure 4. Missing data arises when no pharmacophores occur in 90% of the actives. For the first set of scrambled data (Random 1) the number of false positives is 79 and its is 96 for the second (Random 2). The corresponding true positive hit rates are 36.3% and 37.7%. This marked decrease in the true positive hit rate resulting from the significant increase in the number of false positives suggests that the volume maps are an effective search constraint.
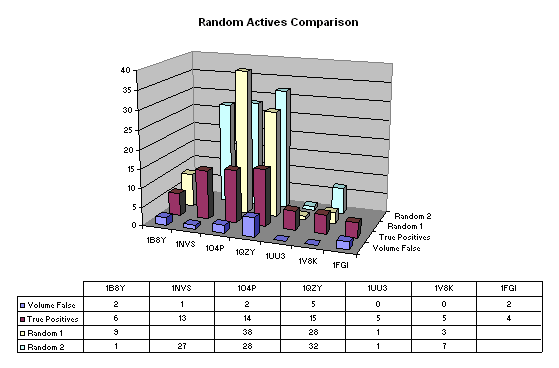
Figure 4: Results from two sets of scrambled data (Random 1 and Random 2) with the 'True Positives' and 'Volume False' positives (see Figure 3) for comparison.
For completeness, we also explored 'leave one out' validation despite the small numbers of molecules and the probability that the model would be unstable. The results are shown graphically in Figure 5. Missing data again arises when there is no common pharmacophore for 90% of the remaining active molecules.
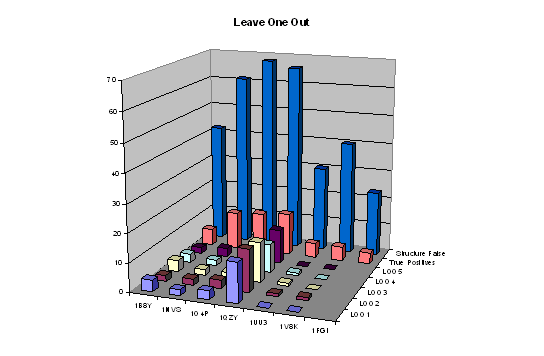
Figure 5: Leave one out validation for five omissions (L001, L002, LOO3, LOO4 and LOO5) with 'Structure False' and 'True Positives' (see figure 3) for comparison.
The total number of false positives for each run omitting different active molecules was 23, 24, 23, 17 and 19. The increase over the 12 found for the full data set reflects the lack of stability of the models - mainly that for 1QZY (although the reason for this is not immediately apparent). These numbers are all substantially lower than those for the scrambled data of 79 and 96 as well as much lower than the number of false positives from structure based virtual screening confirming that the results of the active volume map constraints are significant.
These results show a statistical robustness across a range of proteins that we had not expected. We had no reason to assume that the volume constraint would eliminate many of the false positives found by structure-based virtual screening. However, we can envisage cases when occupying a specific pocket or interacting with certain residues does not contribute much to activity. In order to gain a better understanding of the physical significance of the active volume constraint we constructed union volume maps around the structure-based virtual screening conformers which are predicted to bind. The maps represent the subsite occupied by the conformers. If the false positives were occupying pockets and interacting with residues which are unused by the true positives then we would expect to be able to identify these regions of space and eliminate some false positives. The results are shown in Figure 6.
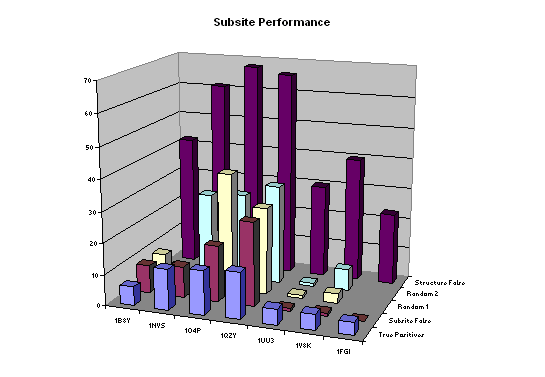
Figure 6: Subsite volume constraints. The 'Subsite False' positives are sometimes fewer than the Random 1 and Random 2 (scrambled data) results. 'Structure False' and 'True Positives' are included for comparison (see earlier figures).
The numbers of subsite false positives are not consistently below random and consequently it is not possible to make the convenient conclusion that structure-based false positives occupy pockets and binding modes that are not used by true positives. Other possibilities include that the active volume constraint is associated with some protein receptor conformation change that is triggered on binding. The fact that a volume constraint is so effective is inconsistent with the popular belief that false positives are associated with approximations in the scoring function.
The method is limited to 3 and 4 centred pharmacophores. If molecules do not bind in the same way then common pharmacophores are not found. In our wider experience we found it was important to avoid assuming all the molecules in the training set would exhibit common pharmacophores. For this data set we found little dependence on which of the smallest 10 volume constraint maps was used and in this example a molecule was a hit if it fitted within any of the constraints. In other cases we have seen greater sensitivity. For highly flexible molecules we found that imposing a limit on the number of conformations that exhibited the pharmacophore reduced the time taken. Based on the results for this data set a limit of 100 would be recommended as 200 gave little improvement and reducing the limit to 50 had a small negative impact for some of the map constraints.
For predictive use a training set is necessary in order to ascertain which pharmacophore and volume constraint is most likely to give the best results. The technology is well suited to lead explosion and lead optimisation but can not be used for lead generation because it relies on an initial set of active molecules.
In conclusion, the volume constraints increased the proportion of true positives from 16% to 84% across a diverse range of proteins. The volume constraint does not appear to be associated with a subset of the initial binding site and suggests that protein conformational change might be important.