
[1]C(=O)Cl + [2]N(H)H > [1]C(=O)N(H)[2]
Figure 1 Example reaction representation of a Combichem Library
E. K. Davies and C. J. Davies
Treweren Consultants Ltd, Holmleigh, Alcester Road, Harvington, Evesham WR11 8HX, UK
The de novo derivative generation and Combichem enumeration functionality of THINK have been enhanced to provide in situ growth of docked fragments to more strongly binding molecules which interact with targeted residues whilst retaining their drug-like characteristics. Critical algorithms have been improved and the number of transformations which can be applied to molecules has been increased. The software integration within THINK allows these poses to be refined with protein side-chain motion and ranked using a scoring function.
Fragment Based Drug Design has for many years been considered the domain of smaller specialist research groups who use protein x-ray crystallography capabilities to locate small molecules bound in protein active sites. Probably the best known is Astex Pharmaceuticals[1], who have pioneered the development of some tools for experimental and computational fragment based approaches. Much emphasis is placed on suitable small fragments [2] using the 'rule-of-three' which constrains the molecular mass (<=300), numbers of Hydrogen bond donors (<=3) and acceptors (<=3), and computed ClogP (<=3.0). Astex and others have demonstrated that traditional medicinal chemistry approaches can use fragments with µM binding to develop higher affinity leads and clinical candidates.
The de novo derivative generation functionality of Treweren Consultant's THINK software was one of the early developments which was used together with the combinatorial enumeration functionality and pharmacophore docking capabilities in Oxford University's distributed computing CAN-DDO project [3] and subsequently in Treweren Consultant's Find-a-Drug project [4]. These projects screened some 3.5 billion drug-like molecules against approximately 300 protein targets. At its peak, the CAN-DDO project used nearly 2 million PCs around the world and Find-a-Drug project generated over 300 million virtual hits (including a relatively small number of 'controls' which had been tested on the NIH cell-based assays).
Both of these projects generated conformations and docked all the molecules independently of successes or failures of similar molecules and experienced a low overall hit rate. Perhaps the most computationally expedient approach would have been to perform the de novo derivative generation in 3D starting with the coordinates of a bound molecule, checking for the additional atoms being in contact with protein atoms and rotating ligand bonds to relieve such contacts. An analogous approach could have been used for 3D combinatorial chemistry enumeration. Both of these approaches have the potential to quickly generate a superset of those which the medicinal chemist might suggest. The approach we have developed for fragment growth also allows protein residues with which additional interactions are desired to be specified and eliminates possibilities which have little potential for better binding with much less effort and time through automation. The pose or starting conformation of the docked molecule can be subsequently refined together with the protein side-chain torsion angles to optimise the geometry of the interactions and improve the ranking of the proposed molecules.
In essence Combinatorial Chemistry enumeration generates structures by combining a core group with one or more lists of R-groups [5]. Some alternatives including constructing chains by connecting R-groups to each other such as in the formation of peptides and using a list of alternative core groups are also possible but not directly relevant to fragment based drug design. When enumerating Combichem libraries within THINK it is usual to use a reaction based representation of the library where the product (or products) represent the core group(s) for the library and the reactants define the require substructures which are used in reagent searches. These reagent searches also convert the reagent molecules into the corresponding R-groups.
|
|
[1]C(=O)Cl + [2]N(H)H > [1]C(=O)N(H)[2] |
Figure 1 Example reaction representation of a Combichem Library |
For Fragment Based Drug Design, the core group is normally a modified form of the bound fragment. A small modification is required in order to convert the fragment into a core group with group connection positions indicating by the numbers 1, 2, 3 etc. THINK includes functionality to interactively edit the molecule in 2D without loss of the 3D coordinates in the reference frame of the protein site. A list of R-groups which might originate from a Reagent Search in THINK is also required, although either the original reagent list or the subsequent R-group list might be filtered to produce a range of groups of an appropriate size, numbers of heteroatoms, molecular mass and other properties.
The enumeration is performed to a 3D SD file using THINK either in native mode or from within the KNIME environment. In the presence of a protein and the bound fragment the software fits the enumerated molecule to the bound fragment by common substructure and randomly selects torsions angles within the R-group to avoid bad heavy atom contacts (less than 60% of the sum of the VdW radii) with protein atoms. Rather than allow any torsion angles, look up tables of regular torsion angles are selected with associated weights for each value to bias the results towards the most common conformers such as extended chain.
The current implementation allows up to 4 lists of residues with which interactions are desired. The randomly generated conformers are allocated a merit score based on the proximity of the atoms in the R-group(s) to the desired residues and is higher for atom pairs with the potential for complementary pharamcophore type interactions. The merit is scaled in the range 0-100 for each residue based on distances between the atom pairs. In the current implementation, the optimum distance between complementary centres such as those forming hydrogen bonds is 3Å which results in a merit score of 100 which reduces on a linear scale to 0 at 8Å. For other atoms, the highest merit score 50 which corresponds to a separation distance of 2Å (the contacts check eliminates lower separation distances). The merit score for each residue is the maximum of that for any of the atom pairs and the overall merit for a conformer is determined by adding together the merit scores for each residue. Thus merit scores of 300 or more are possible providing the numbers of residues in lists is sufficient for 3 or more interactions. The conformer with the highest merit score is retained. (The use of 4 lists is desirable for compatibility with pharmacophore docking and does not impact the merit value).
The de novo derivative generator in THINK uses a table of transformations. On the left of each transformation is a substructure which might occur in a target molecule and on the right is a replacement substructure. In the original implementation, when a randomly selected substructure from those on the left of the transformation table occurred, the 2D and 3D coordinates were invalidated and the connection table modified. The implementation generated further derivatives by modifying those made earlier and allowed the resulting molecules to be rejected based on drug-like properties filters and lists of undesirable, reactive or toxic substructures.
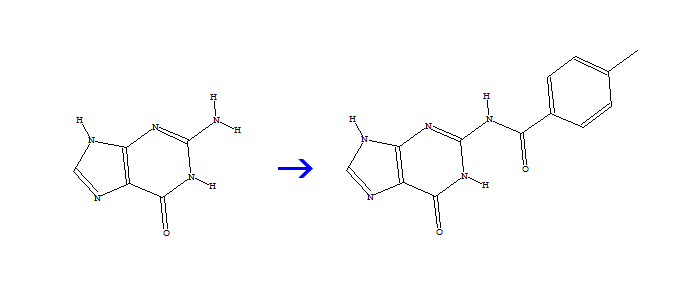
|
Figure 2 Example of a de novo generated derivative |
The approach has been used to generate large numbers of drug-like molecules which the software can attempt to dock into binding sites. However, some of these derivatives can't be docked and others do not generate better scores. Whilst the scope to customise the transformation table, assign probabilities to transformations determining their selection and specifying which parts of the original molecule can modified were useful enhancements, the necessity to generate conformations and dock each new derivatives limited the utility of these capabilities.
The functionality was extended to preserve 3D coordinates for those transformations which performed substitutions, simple chain extensions and reductions requiring only one connection with the static part of the molecule. As a result the 3D geometry can be reliably extended in situ and any clashes between the new atoms and those of the protein identified immediately. The implementation allows the conformation for additional groups to be randomly selected using the same approach as used for R-groups and the same algorithm to compute the merit of each new derivative based on the proximity to any target residues.
For use with Fragment Based Drug Design, the transformation table has been extended to include some which make bigger changes. Most groups which are added by the transformation have been carefully selected from commercially available reagents. Users can restrict the size increase by specifying a curb factor which limits the number of additional non-hydrogen atoms. Above this curb the probability the transformation is reduced linearly until it reaches zero at twice the value of the curb.
Some subtle changes have also been made to improve performance: Transformations which would result in an unacceptable increase in molecular mass are discarded immediately and molecules with no significant merit (ie scoring below 35) are increasingly likely to be randomly discarded as the generations away from the starting molecule increases. In some cases, performance is increased significantly by linking transformations with the same substructure on the left so that by recording substructure search failures, additional searches using the same substructure originating from different transformations are omitted. To assist tracking the ancestry of molecules, the starting molecule and the transformation is recorded for each molecule permitting the genealogy of a molecule to be traced back through generations.
RAS proteins are interesting potential targets for cancer. Cell growth and differentiation appear to be linked with an activated RAS pathway and is found in approximately two thirds of all human cancers. Mutations of RAS occur widely in tumour cells especially in Pancreatic cancer cells. Inhibitors for K-RAS mutant such as Farnesyl thiosalicylic acid as known as Salirasib [6] have shown significant potential in animal studies but not sufficient activity and significant side-effects in (human) phase II clinical trials to warrant further commercial trials. Analogue molecules are being tested and are showing some promise [7].
H-RAS was included as a target in the Find-a-Drug project because it is also associated with cancer and searches were performed using queries based on the PDB structure 821P. In this example, we evaluate the inhibitors grown from the guanine fragment of the bound GTP analogue ligand in 821P by both the Combichem and the De Novo growth software tools. Subject to the default drug-like criteria used by THINK and an atom curb of 6 (see earlier), 2000 derivatives were generated with conformations which didn't clash with the protein which were subsequently refined using the default steepest descent minimisation of the score with respect to the ligand and side-chain torsion angles.
A set of R-groups used in the Find-a-Drug project and originating from 3926 primary halides enumerated to 1671 drug-like molecules. The enumeration used the standard nitrogen connection of the original ligand whilst the de novo generation modifications and connections were unconstrained. In both cases, merit scores were computed based on a list of residues with sequence numbers 14:18 which with which the original ligand interacted. Unlike pharmacophore docking, the refinement is unconstrained to interact with specific residues.
1-494 1-1673  1-1442  1-90  |
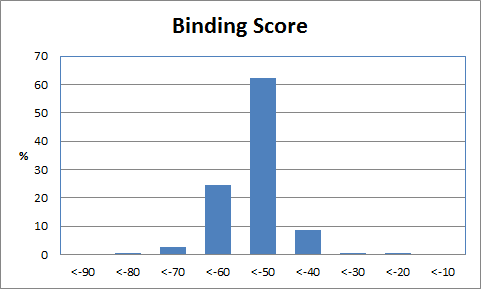 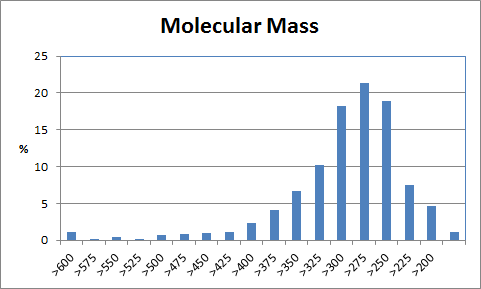 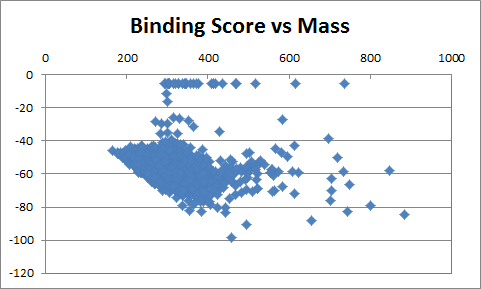 |
| Table 1 Combichem: Representative molecules, Binding Score and Molecular Mass distributions and Binding Score vs Molecular Mass Scatter plot | |
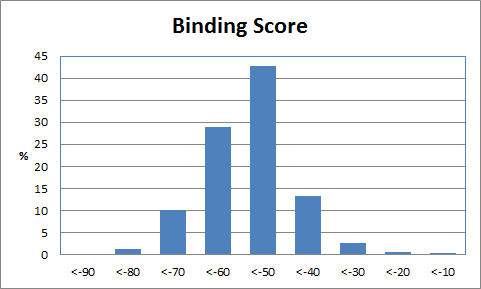
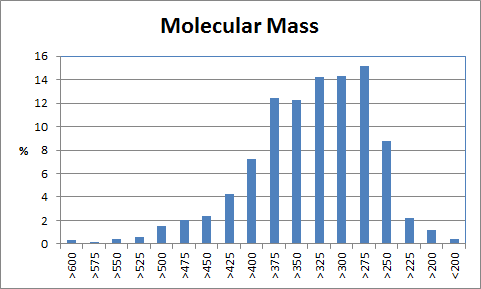
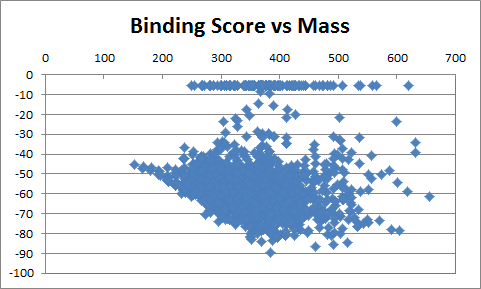
For the Combichem library, a small number of molecules (7 in this library) did not refine because the starting geometry scored too badly and a few more (29) migrated out of the binding site during the unconstrained refinement giving scores of approximately -5. The two best scoring molecules have exceptional scores (less than -90) although the majority are in the -40 to -70 range. The distribution of molecular mass is good with no excessively high values only 18 exceeding 600. Furthermore, there is no obviously relationship between better binding and large molecules. Figure 3 shows the best scoring molecule with the guanine group moved slightly but exhibiting similar interactions to the crystal ligand and the SO3 group positioned close to the original terminal PO3 group.
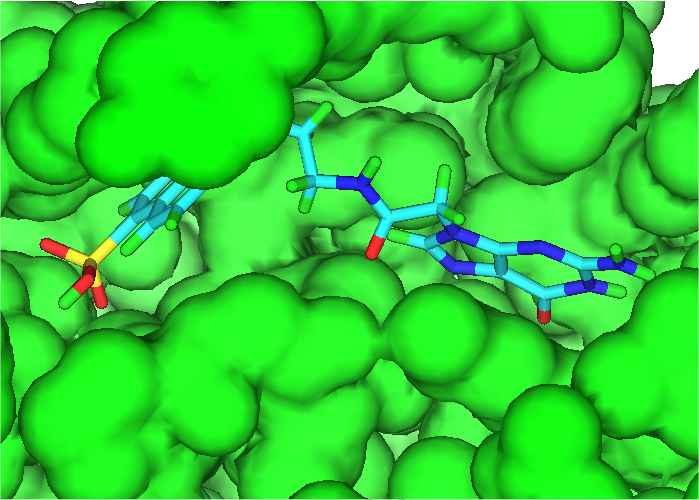 |
| Figure 3 Molecule 1-1503 docked into 821P binding site |
Table 2 shows the corresponding data for the de novo grown molecules which have similar characteristics although the average molecular mass is lower and the best binding scores are less extreme. Of the 2000 compounds docked, 98 had an initial score too high for refinement and a further 301 drifted out of the binding site during unconstrained refinement. This higher number reflects substitutions being made at random positions whereas the Combichem library was limited to the N-substitution found in the crystal structure.
1752 1747  1162  1633  |
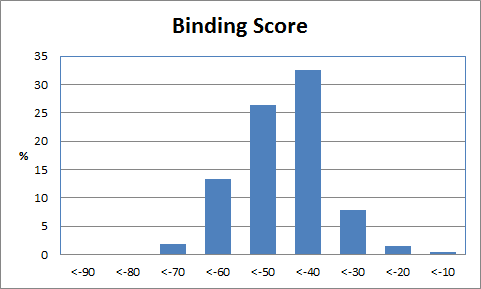 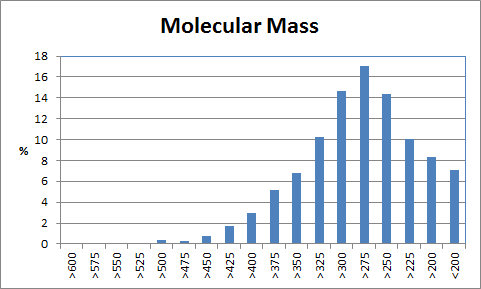 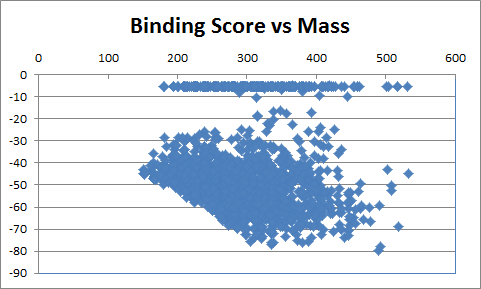 |
| Table 2 Unconstrained de novo: Representative molecules, Binding Score and Molecular Mass distributions and Binding Score vs Molecular Mass Scatter plot | |
When only a subset of atoms in the original fragment are allowed to be substituted, such the nitrogen used for the Combichem library, then the de novo algorithms tend to produce more analogues which differ only by small substitution differences such as halogens. These changes are more likely to be introduced in later derivatives after the scope for adding larger number of atoms using the higher probability transforms at the permitted substitution site(s) are exhausted. In this example, the THINK default drug-like restriction on the number of pharmacophore interaction centres to 9 plays a significant role because the number of nitrogens in the guanine group eliminates some second generation transformations and indirectly limits the sizes of the molecules generated. As the selection of transformations and substitution sites is random, different random seeds, small changes to the transformation table or the drug-like criteria normally result in a different series of molecules being made which may overlap with other series if the number of molecules being made is relatively large. In a restricted de novo generation we generated 2000 derivatives with atom curb set to 8. The number of molecules with an initial binding score too high for refinement drops to 10 and the number of molecules which drift out of the binding site during refinement reduces to 134.
406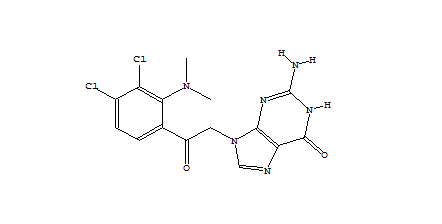 1303 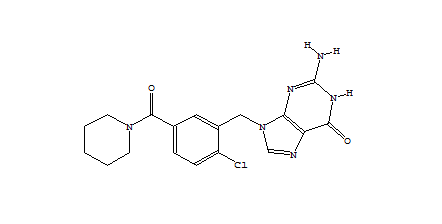 1015 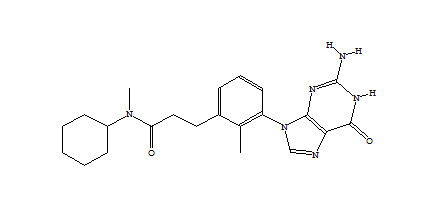 989 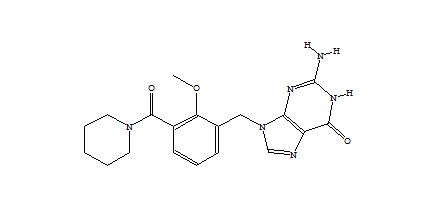 |
   |
| Table 3 Restricted de novo: Representative molecules, Binding Score and Molecular Mass distributions and Binding Score vs Molecular Mass Scatter plot | |
A different starting fragment which preserves the key receptor interactions, such as acetyl-pyridine, could be used with the same R-groups in the Combichem library but would result in a different series of molecules using the de novo approach because different transforms would be applicable. In summary the de novo generation starting from acetyl-pyridine with substitution restricted to the C-5 meta position, generates 25 molecules with extraordinarily good scores below -100. However, with 3 exceptions the molecular mass of these molecules exceeds 500. This is expected because lower number of the number of pharmacophore centres in the starting fragment has allowed substituents to be created with more centres before being eliminated because they are not drug-like. For the Combichem library, although the R-groups are the same, their relative positions especially after relaxing contacts with the protein will be different. For instance, the conformation saved for 1-1503 has a less notable score of -49. Perhaps in the absence of a comprehensive docking with an extensive consideration of ligand conformations, it is reasonable to expect that a significant number of the poorly bound molecules could adopt conformers which bind better.
Both the Combichem and the de novo methods can quickly generate a large number of potential ligands which can then be refined and assessed. Without constraints on the substitution position, most of the de novo generated molecules will not be substituted in the same position as the Combichem library. This means that where there is prior knowledge of the preferred substitutions and R-group sizes, the Combichem approach will generally be more efficient. In some instances, small changes in molecular structure can force a different starting geometry for refinement which contributes to a significantly different binding score.
The use of the THINK software from KNIME allows the refinement to be performed in parallel. Combined with multi-threaded algorithms for especially the calculation of non-bonded interactions, this allows thousands of drug-like derivatives to be refined in a few hours (depending on the speed and number of CPUs). The time taken for refinement can be reduced by using rigid side-chain conformations in the protein but this is also decreases the range of binding scores and often results in molecules with strong binding potential being missed.
As the Combichem library uses commercially available reagents for the R-groups, their novelty and the scope for occupying unique chemical space are intrinsically dependent on the ubiquity of the reagents. Consequently, the de novo grown molecules might have a greater intellectual property potential especially if proprietary fragments and transformations are used.
One might expect some of the larger de novo grown molecules to be synthetically less tractable than typical Combichem generated molecules of the same size but no detailed analysis of synthetic accessibility is included this study. There is potential for alternative starting fragment and substitution patterns to have an impact on synthetic accessibility and binding potential. The effect of changes to the starting fragment suggests that these fragment growth approaches are perhaps better at providing ideas than predictions unless the molecules are re-docked with a comprehensive exploration of conformations - which is time-consuming!