 |
THINK using KNIME |
 |
|
| |
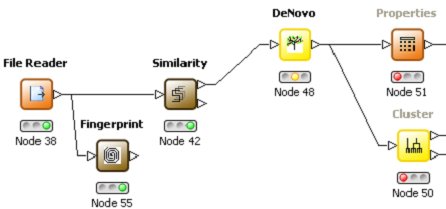 |
Developed by the University of Konstanz KNIME (pronounced naim) provides a means of visualising and configuring the steps for a molecular modelling study where each step has a pictorial representation as a node in a workflow. Using workflows for computer aided chemistry is both easier and more intuitive than traditional GUI and command scripts. Workflow technology is well established in the pharmaceutical industry for ChemInformatics applications usually processing 2D information, for instance to select subsets of commercial catalogues. THINK includes a range of 2D functionality some of which revolves around 2D functional group keys. These can be used to generate molecular fingerprints and also for similarity searching and clustering. The fingerprints can of course be analyzed and manipulated by standard KNIME nodes. The range of THINK functionality provided includes the popular de novo derivative capabilites to generate drug-like analogues of existing molecules. Integration into KNIME and the use of de facto molecular data standards such as SMILES and SD format allows its full range of statistical tools to be used with molecular properties calculated by THINK and other third party software. |
| |
|
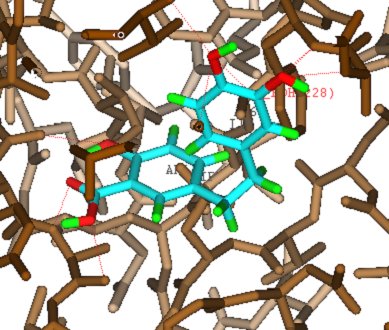
THINK is probably best known for its Structure-based virtual screening capabilities because it was used in two major Distributed Computing projects: the Oxford University Cancer project and the Find-a-Drug project.
Pharmacophore technology is used matching interaction centres in potential ligands to possible interactions with the protein residues. A full conformational generation is implemented and an enhanced ChemScore function is used to score the hits. A comprehensive implementation includes options for the user to elect to refine side-chain positions, save all docked conformers, visualise the hits etc. | 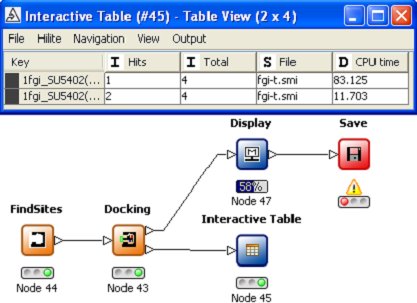
Probably one of the most exciting aspects of using KNIME is the ready automation of tasks which were previously considered tedious and time-consuming. For instance, if there are several possible binding sites THINK can automatical locate these from bound ligands; PDB site records or a site search and dock a set of training molecules into each site summarizing the results in a table. This enables higher quality modelling studies - optimising the configuration for a training set - to be completed with less effort! Note:The molecular graphics THINK nodes are currently only released for Windows. There are third party molecular graphics nodes for Linux. |
| |
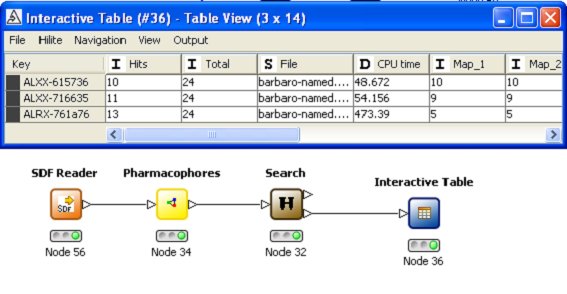
The computation of common pharmacophores and the associated map volume constraints is explained elsewhere. However using KNIME, the workflow is visually communicated and the performance of various alternative common pharmacophores and associated map constraints is presented as a table. This allows rapid assimulation of how well the technology is suited to a particular problem. |
The pharmacophore technology originally pioneered in Chemical Design's Chem-X software has now been extended and is available using KNIME. A recent and valuable extension is the use of volume constraints with pharmacophores which has unpresidented accuracy predicting which molecules will be active - over 80% true positives.
|
| |
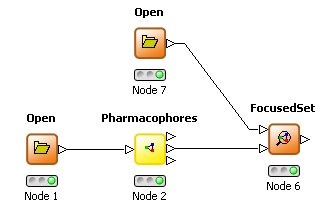 |
The implementation of the pharmacophore node for KNIME was enhanced in version THINK 1.41 to provide more ready access to the pharmacophore technology for designing diverse and focused libraries. The pharmacophore output has the pharmacophores for each molecule and is designed to be used with standard KNIME nodes and statistical tools. The profile output is the average list of pharmacophores for the set of molecules and this can be used for selecting molecules. The FocusedSet node selects a subset of the molecules which exhibit pharmacophores which overlap with those in the profile. The degree of overlap required may be adjusted in the configuration dialog. The DiverseSet node provides the complementary functionality by selecting molecules which exhibit additional pharmacophores. |
| |
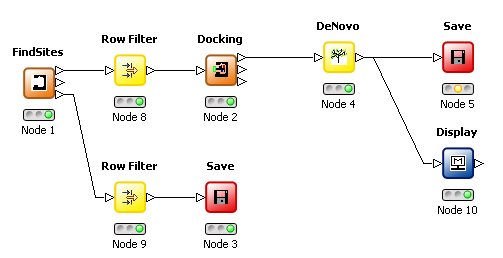 |
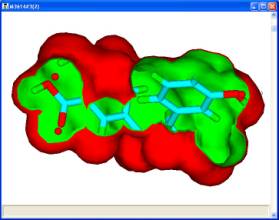
The De Novo node can suggest and evaluate derivatives of known ligands which have been docked into a binding site. The transformations used are limited to those which perform substitutions so that the core of the molecule and its interactions are maintained. Derivatives which overlap with protein atoms are rejected. If derivatives of the ligand observed in a crystal structure are required, then the third output of the FindSites node allow this molecule to be saved prior to being processed by the Docking node. |
This technology can also be used to generate derivatives of hits from a pharmacophore search with map constraints. |
 |
|