
This manual is designed to provide supplementary information on selected areas of THINK for the more expert user who wishes to understand some of the algorithms used. It is not intended to provide details on how the program is used that is the function of the User Guide. In some circumstances it will be necessary to contact Treweren Consultants, read the source code or the scientific literature in order to understand certain algorithms and implementation issues.
The manual is divided into chapters that cover
and includes details of some file formats and common variables that can be examined by the user.
2 Reading and Saving Files
THINK reads and writes small molecule connection tables in the following formats:
The PDB format is designed for use with proteins and peptides. In this manual, the term SDF file will be used to refer to both molfile and SDfile formats.
Although THINK uses both 2D and 3D coordinates, it is not necessary for these to be included in the SMILES or SDF file. If the appropriate coordinates are not available, THINK will transparently generate 2D coordinates when required for display, and 3D coordinates when required for display, property calculation or conformational analysis.
The molecules in a combinatorial library may be automatically enumerated as they are saved to a file. This chapter includes a section on enumeration.
THINK also provides the ability to read and write selected parameter files, namely atom types, bond and angle parameters, ring coordinates and torsion angles (for use in conformational analysis).
File names which contain spaces should be enclosed in double quotes in scripts or when entered as a command line. File names should not start with "+" or "-" as these are special characters that force an append to or replacement of an existing file which can be useful in scripts.
2.1 SMILES
The SMILES format was developed by Daylight Chemical Information Systems, Inc (www.daylight.com/smiles) and is a compact line notation that uses a hydrogen-omitted connection table. Blank lines and lines beginning with ! are ignored. A full description can be found on their website. The format has been extended for THINK to handle molecule names and field data.
2.1.1 Key Features
This file uses one line for each structure. For the common elements of organic molecules (C, N, O, S, P, B, F, Cl, Br, I), known as the basic SMILES elements, the element symbol is used to indicate its presence with parenthesis for indicating branches:
eg CCC(=O)O
This example also indicates the use of = to signify a double bond. Triple bonds are shown by # and, where necessary, single bonds by - and aromatic bonds by :. For delocalised bonds, ~ has been reserved but is not generally supported by THINK. Lowercase letters are expected for atoms in aromatic rings, when aromatic bonds are assumed, although Kekule representations may also be used.
Elements other than the basic SMILES elements should be enclosed within square brackets []. Brackets also required if a formal charge or explicit hydrogen count is used:
eg [O-2] or C[NH2+]C
In such cases, where the value exceeds unity an integer must follow the +, - or H. A ring closure bond is indicated by an integer after the pairs of elements being bonded:
eg c1ccccc1
When a ring closure is used with square brackets, the ring closure digit(s) follow the ].
2.1.2 Extensions
Atom types instead of element symbols may be used inside square brackets. Where these have the potential for conflict with other SMILES representations, they should also be enclosed within double quotes :
eg [CSP3] or [N+]
For combinatorial chemistry, the ability to store R-groups and define R-group queries is important. The atom type 0 is used for a generic connection atom and types 1-9 are used for explicit connection atoms (see section 2.4). Since these are non-standard atom types, they must be enclosed in square brackets [] when used in SMILES files.
For R-group queries (see section 8.3), the < is used between the atoms which are to be retained as the R-group and those which are discarded eg C1<N1([H])[H] which would generate R-groups from primary amines with a generic connection atom (atom type 0) replacing the nitrogen. Note that in these circumstances it is important to specify the hydrogens connected to the nitrogen. Bonds between the two sets of atoms are shown using ring closure digits.
The group number for disconnected fragments may be specified by an integer (in the range 0-9) immediately following the .. This is rather useful when defining queries for R-group searches eg C1.3C2<C1N12[H] that will generate two R-groups from secondary amines.
Numeric field values may be stored on the same line after the SMILES string in the form:
field=value
where field is the name of the field and value is a real number or integer. Note that there are no spaces around the =. The reserved field NAME is used to indicate the molecule name. (THINK v1.25 does not support text variables).
2.1.3 Limitations
There is no support for storing atom coordinates.
2.2 Molfile and SDfile
These formats were created by MDL Information Systems, Inc (www.mdli.com) and are widely used by various programs. A full description can be found on their website.
Although THINK can read both molfiles and SDfiles, it can only write SDfiles.
2.2.1 Key Features
The heart of a molecule definition within a molfile or SDfile is the Ctab connection table block. This is a fixed-format block of records containing information about the atoms, bonds and properties (eg formal charges) of the molecule.
A molfile consists of a Ctab block preceded by three header lines, and stores a single molecule. The molecule name may be stored in the first header record, or in the second header record in place of the molecules internal registry number (starting in column 47). If the name occurs in both records, the name from the second record will be given priority. The CUSTOMISE NAME=field command allows this priority to be reversed or a field to be used for the molecule name.
An SDfile consists of multiple molfile blocks, each followed by a record containing $$$$. Any field data associated with a molecule is placed between the molfile block and the terminating $$$$ record. The data for each field must be preceded by a record starting with > and containing the name of the field enclosed in angle brackets <>.
THINK uses the following FORTRAN format to store the atom data:
1 2D for 2D files; 3D for 3D files. An extension, also used by Chem-X, allows both 2D and 3D coordinates to be stored within the atom records in the Ctab block. The second set of coordinates is placed in columns 76-105 using the format 3F10.4
2 Element symbol for normal atoms; atom type name for dummy atoms. For dummy atom types with 4-character names (eg AROM, BASE), the 4th character is written into the preceding blank space
3 Code indicating formal charge: 4-q, where q is the formal charge
4 Code indicating chiral parity
5 Code indicating hydrogen count: nh+1, where nh is the number of missing hydrogen atoms
6 The atom mapping number is used as the serial number (if non-zero) and if necessary is allowed to exceed the 3 allocated characters to overwrite the reactant/product field. The reaction component type (reactant, product or reagent) is used for the group number
The extensions allow SDfiles to be used to store R-groups and R-group queries, although the more compact SMILES format would normally be used. However, THINK v1.25 cannot save R-group queries in a SMILES format file.
2.2.3 Limitations
The query constructs used by Chem-X, ISIS and other software are not currently supported.
THINK v1.25 does not support the use of text fields.
2.3 PDB Format
The PDB format is the de facto standard for 3D protein coordinates. Unlike MDL SDfiles, the amino acid or residue information is included. See www.rcsb.org/pdb for further information on this format.
2.3.1 Key Features
A subset of the standard keywords are processed:
| Keyword | Description |
| ATOM | Atoms with standard amino acid residue names |
| HETATM | Atoms in non-standard residues |
| CONECT | Connectivity records for non-standard residues |
| COMPND | For molecule name |
| HET | To identify non-standard residues |
| HETNAM | Names of non-standard residues |
| HETSYN | Synonyms for names of non-standard residues |
| HELIX SHEET TURN |
Sets of residues comprising secondary structures |
| SITE | List of residues comprising an important site in the molecule (eg active site) |
An additional keyword, NAME, is also interpreted. This is an extension used by THINK, and must be manually inserted into the PDB file using a text editor (see section 2.3.2 below).
Unlike SDF and SMILES format, hydrogens are not automatically added unless the HYDROGENS option is specified.
A symbol is automatically created for each helix, sheet strand or turn defined in the PDB file. The symbol contains the range of residues that define the secondary structure unit, identified by their sequence numbers and, where necessary, the chain ID eg (124:127) or (266:272)B. The symbol name has the form HELIXnn-dd, SHEETnn-dd or TURNnn-dd, where nn is the number associated with the secondary structure (read from columns 8:10) and dd is the secondary structure's identifier (read from columns 12:14).
THINK will automatically create a symbol for each site definition found in the file. The symbol consists of an array called SITE-xxx where xxx is the site identifier (read from columns 12:14 of the SITE record). Each residue in the site is identified by its name, sequence number and, where necessary, chain ID and is placed in a separate array element.
The NAME keyword is used to define the molecule names. NAME records are added to the PDB file using a text editor. If used, a separate NAME record must be inserted at the beginning of every molecule in the file. If a molecule has any CONECT records, these should appear in the same section of the file as the molecule's ATOM or HETATM records (ie before the NAME record for the next molecule). This may require moving some of the CONECT records.
A NAME record should contain the keyword NAME in columns 1:6 and the molecule name in columns 7 onwards.
Older versions of THINK (v1.12 and earlier) would read the molecule name from the COMPND record. This has been superseded by the introduction of the new format of COMPND records with version 2.0 of the PDB format (see www.rcsb.org/pdb and Molecule Names below).
Atom radii may be read from and written to the temperature or B-Factor field (columns 61:66) in ATOM and HETATM records. This may be useful to set an atomic tolerance for site search queries.
Atom group numbers are stored in columns 67:69 in ATOM and HETATM records.
2.3.3 Molecules and Molecule Names
If NAME records have been added to the PDB file, they will be used to define the molecule names. In the absence of NAME records THINK will take the molecule names from the following records:
| Keyword | Columns in record |
Description |
| HEADER | 63:66 | PDB protein ID code |
| COMPND MOLECULE: | 20:70 | Macromolecule name |
| COMPND SYNONYM: | 19:70 | Synonym for macromolecule name |
| COMPND EC: | 14:70 | Enzyme Commision number associated with molecule |
| HET | 31:70 | Description of the HET group |
| HETNAM | 16:70 | Chemical name of HET group |
| HETSYN | 16:70 | Chemical name of HET group |
Continuation COMPND records will be ignored, and only the first MOLECULE, SYNONYM or EC record will be used.
THINK will automatically create a separate molecule for each ligand, and a single molecule containing all the water molecules in the PDB file providing there is a HET record for each ligand and there are no NAME records in the file. The name of each ligand molecule will be taken from the HET record or its associated HETNAM or HETSYN records. The water molecule will be called xxxx-WATER where xxxx is the PDB protein ID code. If the file contains two or more ligands with identical names, THINK will add a counter of the form "(n)" to the molecule name to distinguish the molecules.
It is recommended that NAME records are used when the ligand binding uses one or more water molecules. The water molecules involved should be included as part of the protein when constructing a site query. NAME records should also be used when the ligand is a peptide (since it will not appear in a HET group so THINK will not detect it as a separate molecule unless a NAME record is used).
2.3.4 Limitations
Connectivity and bond order is deduced from the amino acid residue names and atom names with the consequence that the use of non-standard amino acids (including nucleic acids) or atom names will result in the connectivity being incorrect. Although THINK processes the CONECT records in the file, these do not include bond-order and THINK does not automatically generate this information (for instance from the inter-atomic distances). The command MODIFY REBUILD=CONNECTIONS can be used to regenerate the connectivity, but this deduces the bonds and bond order from the atom coordinates, and therefore will only give the correct connectivity if the atom geometries are accurate.
Normally the molecules saved to a file are identical to those in memory. To generate and save enumerated molecules it is first necessary to read all the R-groups into memory (including any core or central group). The enumeration code permutes the molecules which have been read from the specified files, pairing the explicit connection atoms (those with atom types 1-9) and converting generic connection atoms (those with atom type 0) to explicit connections according to various rules.
There are three conceptually different types of library that might be built:
When generic reaction has been used to search for R-groups, then the product(s) are the core group(s) and can be used during the enumeration. Alternatively, a SMILES file containing just the core group(s) can be created with explicit connection atoms (using atom types 1-n, n £ 9). When creating R-group files (see section 8.3), it is important to use file name conventions that avoid confusing the reagents and the various R-groups that can be derived from them. The scope for confusion is greatest when more than one R-group may be derived from the same reagent.
The R-groups are grouped according to the file from which they were read. The order of the R-groups is specified as part of the enumeration instructions issued when the molecules are saved. Regardless of the type of library being built, THINK will always treat the first file specified as containing the core group. The R-groups from the second file will be connected to the core group; those from the third file etc may be connected to the core or to the preceding R-groups, depending upon the type of library.
The following rules governing the conversion of generic to explicit connections may be useful when attempting to locate errors in R-groups, or to deduce possible library representations:
Errors will be reported for R-groups with more than two generic connections, or if there are unpaired connection atoms (except for the first and last connection atom when building peptide chains).
2.4.1 Core-based Libraries
In order to use the same file of R-groups in several different libraries, the concept of a generic connection (atom type 0) is available. Each R-group should only contain one generic connection and it is critical to specify the R-groups in the correct order following the core group in the enumeration command. If an R-group has more than one connection to the core it is advisable to use explicit connections, and often easier to place such groups as the last R-group. When a mixture of explicit and generic connections are used in R-groups, then each generic connection is converted to the next unused connection taken in the order of the R-groups (and this may leave gaps).
In principle, the core may also use generic connections when they are converted into connection atoms 1-9 based on the order in which they occur. As this introduces scope for error it is not recommended; instead the core group should have explicit connection atoms with types 1-9 referring to the R-groups
2.4.2 Coreless Libraries
Many simple two-component libraries such as amides can be represented without a core. When using THINK, either group may be specified as the core and the other group as the first R-group. Both groups may have a single generic connections or explicit connections (which must match).
2.4.3 Peptide Chains
Although peptide libraries can be represented as a backbone core with the amino acid side-chains as the R-groups, it is often more convenient to use the backbone and side- chain as a building block with two connections. For this to be used in all positions (including the core) it must have two generic connections eg [0]NC(C)C(=O)[0] and it is usually appropriate to use a capping group (eg OH) at the end of the chain as the last R-group.
The implementation includes two important but subtle rules
The connections are numbered according to the order in which they occur within the R-group. It is important that the amino acid R-groups are specified in a consistent direction problems will arise if some are specified as N to C and others as C to N.
2.5 Parameter Files
THINK includes the ability to read and write the following parameter files:
All the files use fixed formats, as described in Appendix A.
If the user wishes to make changes to the parameters, it is envisaged that the existing data will first be saved to a file, which is then edited before being read back into THINK. This will help to ensure that the correct data and formatting is supplied.
3.1 2D Calculations
3.1.1 Counters
The counters that may be evaluated are summarised below:
| Atoms | Positive charge centres |
| Bonds | Negative charge centres |
| Heteroatoms | Rings |
| Branches | Aromatic rings |
| H-bond donors | Heteroaromatic rings |
| H-bond acceptors | Halogens |
| Centres |
3.1.2 Volume and Surface Area
The volume and area of a molecule are estimated using standard bond lengths and radii, without generating 3D coordinates for the atoms. The volume of each atom of radius R is calculated as a sphere 4pR³/3 and the overlap volume with the spheres for connected atoms ph(h²+3r²)/6 is subtracted as shown.
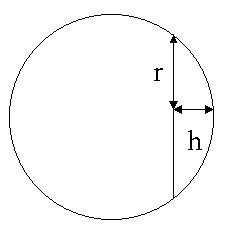
The surface area of each atom is calculated as a sphere 4pR² and the overlap area with the spheres for connected atoms 2phr is subtracted.
3.1.3 Partial Surface Areas
In addition to the total surface area, a series of partial surface areas are calculated:
| Name | Description |
| PSA | Polar surface area |
| NPSA | Non-polar surface area |
| PFA | Polar fractional area (PSA/area) |
| NPFA | Non-polar fractional area (NPSA/area) |
| XSA | Nitrogen and oxygen (plus attached hydrogens) surface area |
| XFA | Nitrogen and oxygen (plus attached hydrogens) fractional area (XSA/area) |
In these calculations, atoms that are hydrophobic (see below) are considered non-polar. Values of XSA above 120 are sometimes used to eliminate molecules that are expected to be poorly absorbed, and only those with values below 70 are commonly expected to penetrate the blood-brain barrier.
3.1.4 Flexibility
The flexibility is computed by the product of the number of increments for each rotatable bond (ignoring ring bonds):
![]()
where Pn is the number of points or increments for bond n. By default, P is 3 except for conjugated bonds where P is 2. Note that this computation is independent of the algorithm used by conformational analysis for detecting rotatable bonds, and is not affected by the corresponding settings.
3.1.5 Lipophilicity
This provides a measure of lipophilicity of a molecule in the range 0-1 using the following algorithm:
This algorithm is also used to create lipophilic dummy centroids or lipophiles when molecules are read into THINK (they cannot be created at any other time). These have the element symbol LIP and may be defined by a minimum of 3 and a maximum of 8 connected hydrophobic atoms. The lipophile calculation:
Note: While this algorithm creates separate lipophiles for fused rings, connected hydrophobic atoms which span fused rings cannot form a single lipophile, even if each ring contains fewer than 3 non-fused atoms.
3.2 3D Calculations
These calculations require 3D coordinates, either read from a PDB or 3D SDF file, or generated automatically by THINK.
3.2.1 CPK Contacts
This calculation reports the number of atom pairs separated by less than the sum of their CPK radii, excluding those that are 1,2 connected (bonded), 1,3 connected (forming a bond angle) or 1,4 connected (forming a torsion angle). The CPK radii are computed by multiplying the VdW radii by a user-defined factor, normally 0.6.
3.2.2 VdW Contacts
This calculation reports the number of atom pairs which are not 1,2 connected, 1,3 connected or 1,4 connected, with a separation distance less than the sum of their VdW radii.
An external program may be used to calculate one or more user-defined properties for a molecule. The molecules connection table is passed to the program via a SMILES or SD file, and the property values are returned via a data file. Use of the external program is enabled by setting the common variable IREMOT to 2 if a SMILES file is to be used, or 4 for an SD file (the default value of 0 means that the external program is disabled). The name of the program is stored in the REMOTE symbol:
| LET #IREMOT = 2 LET REMOTE = program |
As THINK calculates the properties for a molecule, it will automatically run the external program, passing the name of the connection table file (molecule.smi or molecule.sdf) as an argument. The program should write the calculated property values to a text file molecule.dat using one or more records of the form:
| field=value | |
| or | field=value, field=value, |
where field is the name of the field, and value is the calculated value.
If the values are pre-calculated (eg taken from the literature), the same technique can be used to load them into THINK, but there is no need to use a real external program the REMOTE symbol may be set to any arbitrary string. In these circumstances, THINK will just read the field names and values from the file molecule.dat.
Conformational generation requires 3D coordinates. These will be generated automatically by THINK if they have not been read in from a molecule file.
The conformational analysis implementation automatically detects rotatable bonds for the following classes of bonds:
and considers the specified number of positions (points) about each class of bond by incrementing the torsion angle about the bond. If zero positions are specified then all bonds of that class are ignored. The number of points about each class of bonds can be altered though the CUSTOMISE command.
THINK automatically applies an offset of 50% of the increment to each alpha bond during a systematic search. For instance, if the increment for alpha bonds is 60°, each alpha bond will be rotated by 30°, 90°, 150°, 210°, 270° and 330°. For conjugated bonds in sterically hindered environments, THINK will apply an offset of 50% of the increment before starting to rotate the bond thus sampling 90° and -90°. Crowded bonds are not sufficiently hindered conjugated bonds to ensure that they are non-planer. Sampling these bonds at 0°, 90°, 180° and -90° is often best.
The value for ring bonds is the maximum number of ring conformations to be considered, not the number of points for each ring bond. The torsion angles for ring conformations are stored in a lookup table. If the number of ring conformers required is less than the number stored then the lowest energy conformers are used.
During 3D and site searches (see sections 8.4 and 8.5 respectively), it may be desirable to limit the number of conformations generated for each molecule, and to ignore extremely flexible molecules, to prevent the search taking too long to complete. The common variable RCNFMX, when given a non-zero value, sets the upper limit for the number of conformations (a value of zero means that there is no limit). When a molecule is first scanned to detect the rotatable bonds, THINK automatically computes the total number of conformations that will be generated. If this exceeds the value of RCNFMX, each single (sp³-sp³) or alpha bond that has 12 points is reduced to 6 points until the total falls below RCNFMX. If necessary, the process is repeated for the single bonds, reducing the number of points to 3. If this still fails to bring the total below RCNFMX, the molecule is ignored.
4.2 Conformer Generation
The systematic search option sequentially increments the torsion angles about each rotatable bond in turn. This can be very time-consuming for flexible molecules, because it is comprehensive, but may be accelerated using the contacts check (see below). In addition, the conformational search may be abandoned after a certain time interval, specified through the CUSTOMISE TIMEOUT keyword. This is particularly useful in 3D and site searches (see sections 8.4 and 8.5) to prevent THINK spending a long time searching a single molecule.
Random sampling makes an estimate of the total number of conformations (computed by the product of the number of increments about each bond) and then uses a random number generator to select conformations randomly from within the estimated total. The implementation does not prohibit the same conformation being selected multiple times, and this will always occur if more conformations are requested than the total number of different conformers.
Random sampling does not necessarily produce the most representative set of conformers. A further sampling option is available, which attempts to space the sample conformations uniformly across conformational space. If conformations are rejected (for instance because of VdW contacts), then up to 10 attempts are permitted in order to find a sample interval which will explore additional conformations. The implementation ensures that the same conformation is not repeatedly sampled. However, this does not prohibit identical conformations to be output resulting from symmetry.
4.3 Contacts Check
In order to prevent high energy conformers being considered, it is necessary to use the VdW or CPK contacts check. For systematic search, this also significantly reduces the time taken because when atoms in contact are detected, rotations about bonds which do not change the inter-atomic distance are skipped. There is no acceleration for either sampling method.
5 Data Analysis
The data analysis is designed for interpreting HTS data and ADME-Tox data by extracting features which distinguish inactive molecules that show undesirable activity from active molecules. These features may be properties calculated by THINK, functional groups used in the functional group keys or pharmacophores. The implementation uses a form of discriminant analysis appropriate for the types and distribution of data (which is not usually a statistical normal distribution). This technique is appropriate for up to 10,000 molecules except when pharmacophore option is used when memory limitations usually restrict the analysis to a maximum of about 1,000 molecules. Larger numbers of pharmacophores can be imported and analysed in relational databases.
The pharmacophores exhibited by each molecule must be calculated in advance of the data analysis and stored in a file named molecule.phm where molecule is the name of the molecule. It is recommended that the SAVE OPTIONS=FILTER command is used when generating the pharmacophore files to prevent files being created for very flexible molecules. It is also recommended that all salts are removed from the structures before the pharmacophores are calculated, since salts may contain centres (eg oxygen atoms) which would lead to the creation of additional pharmacophores partially or wholly outside the molecule that is to be used in the data analysis. This most conveniently executed as part of a script which loops over all molecules:
| OPEN FILE='P1' |
| JMOL = 0 |
| WHILE JMOL < #NMOLES |
| JMOL = JMOL + 1 |
| MOL = $MOLECULES(JMOL) |
| Message = "Processing: " . MOL |
| WRITE CONSOLE Message |
| FILEOUT = "-" . MOL . ".phm" |
| SAVE FILE='FILEOUT' MOLECULE='MOL' OPTIONS=FILTER |
| END |
The results of the analysis are stored in a learn file (with a .lrn extension) which contains the field name undesirable substructures, the F-test value, the acceptable ranges and the confidence limit for each feature. The headers or first line in the following example is NOT included in the file.
| Functional group or field name | F-test | Min | Max | Confidence | Comment |
| ZC(=O)OH | 21.102 | 0.5 | 1.0E35 | 99.51 | (203/962:0/32) |
| OCCN | 12.162 | 0.5 | 1.0E35 | 99.153 | (117/759:0/32) |
| [CAR]OH | 10.499 | 0.5 | 1.0E35 | 99.02 | (101/642:0/32) |
| Branches | 6.42 | 7 | 1.0E35 | 98.974 | (193/541:1/32) |
Learn files created by the data analysis include a comment (Is/It:As/At) where Is and As are the number of inactive and active molecules respectively selected by the feature, and It and At are the total number of inactive and active molecules.
Learn files can be used as rejection criteria in de novo molecule generation or for highlighting rejected molecules in the spreadsheet. They can also be used to filter the molecules saved to a file or accepted during a search. In general, they have the same name as the activity field with which they are associated. THINK v1.25 does not support the use of learn files when selecting subsets of molecules based on property diversity.
5.1 Key Features
The algorithms used have been optimised to extract features that may be associated with undesirable activity, with the consequence that it is important to specify whether desirable activity increases or decreases as the values stored in the activity field increase. In addition, because HTS data is often unreliable, THINK may be instructed to take only the significantly active and inactive molecules, rather than simply dividing the compounds into active and inactives. This is done by specifying a significance value in the range 0-0.5, which selects only the most and least active molecules for the analysis and discards those in a mid-range grey area. A significance value of 0.5 would simply divide the activity range at the mid-point for actives and inactive molecules, whereas 0.3 would take the top and bottom 30% of the compounds.
5.2 F-test for Properties
For properties, the active molecules may be divided from the inactive molecules by a single value (as shown in Case I), or may be bracketed by a pair of values (Case II).
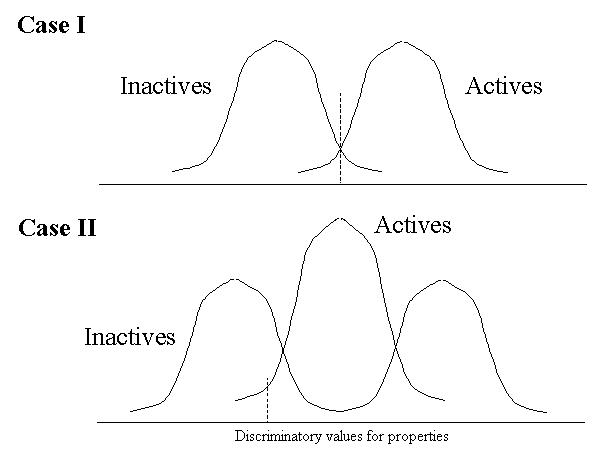
THINK assumes a normal data distribution to estimate these cutoff values for all the properties. The F-test for each feature (which is classically the variance explained by the feature divided by the unexplained variance) is calculated based on the actual discrimination achieved, according to the following formula:
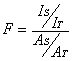
where IS and AS are the number of inactive and active molecules respectively selected by the cutoff value or range, and IT and AT are the total number of inactive and active molecules.
5.3 F-test for Functional Groups
For functional group-based discrimination, THINK determines whether a functional group occurs more frequently in the actives or inactives and hence whether it is desirable or undesirable. The actual discrimination achieved by the functional group is determined in an analogous manner to that achieved by properties.
5.4 F-test for Pharmacophores
The number of times each pharmacophore is exhibited is used in the data analysis in the same way as the numerical properties such as the count of the number of rings. The number of pharmacophores being processed is often very large and consequently the analysis is significantly slower than those which use just properties and/or functional group keys.
5.5 Prediction
Discriminating features are iteratively extracted in order of decreasing F-test result. The confidence and risk for the feature are computed using the following formulae, which are based on a worst-case application of Bayes theorem:
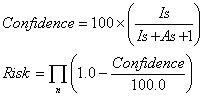
where IS and AS are the number of inactive and active molecules respectively selected by the feature. Note that this analysis approach is predicting inactivity, not activity, hence a large statistical confidence value is associated with inactivity and a low risk value with an undesirable (low activity) molecule.
6 De Novo Structure Generation
The de novo structure generator starts with a specific molecule and generates analogues by applying transformations. Each resulting molecule is tested for property and substructure rejection criteria, and for similarity to active and inactive molecules prior to an annealing step which determines whether the molecule is saved in a file and used for the next iteration. This scheme is summarized below:
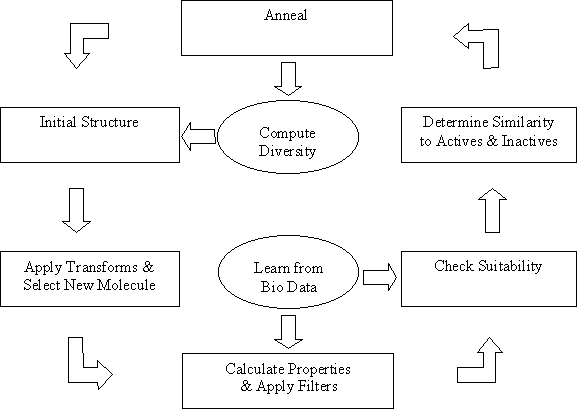
The transformations are stored in SMILES format by default in tranform.smi in the THINK_EXEC directory. One of these is selected at random for application to the molecule; if no substitution exists for this transformation then an alternative random selection is made. If the transformation could be applied at more than one location within the molecule then a site is selected at random. The transform number used is stored in the field TRANSFORM for the resulting molecule.
A user-defined transformation file may be used instead of the standard file. Each transformation is defined in a single record, using the SMILES format to describe the original and transformed substructures with a > as the separator: eg C(=O)C>C(=O)OC would describe a transformation that inserts an oxygen atom. Where necessary, hydrogen atoms must be explicitly specified within the original and transformed substructures.
The selection of transformations can be influenced by defining a probability (which is similar to a relative reaction rate) for each transformation in the file. The values are stored in a field called PROBABILITY. The chance of a particular transformation a being selected is Pa/Psum where Pa is the probability of transformation a and Psum is the sum of all the probabilities in the file.
6.2 Application of Earlier Results
When selections of active and inactive molecules are available, these may be used in two ways:
The file of molecules to be compared must contain activity data. By default, the file is expected to have the same filename as the activity field (as would normally be the case if the file contained results from an HTS assay). The corresponding learn file will be loaded if it has the same file name except for the .lrn extension. When no comparison file is specified, the file default.lrn from the THINK_EXEC directory will be applied. If the learn file cannot be opened then default ranges of heteroatoms (2:9), molecular mass (175:800) and lipophilicity (0.28:0.72) will be used.
6.3 Rejections File
A default rejection file is provided (reject.smi in the THINK_EXEC directory)
which contains some substructures that are known to be unstable, readily metabolised
or impossible to synthesize. This file uses the SMILES format. A user-defined
rejections file may be used instead. Each substructure must be defined in a separate
record, using the SMILES format. Comments may be included in the file providing
they are contained within records that begin with !.
7 Pharmacophores
THINK can calculate 2-, 3- and 4-centre pharmacophores, though most users would normally use only 4-centre pharmacophores as these provide far more shape information and resolution.
The following centre types are recognised and used in pharmacophores:
| Centre name | 1-letter code | Description |
| HDON | D | Hydrogen bond donor (may require an attached hydrogen atom) |
| HACC | A | Hydrogen bond acceptor |
| POS | P | Positive charge |
| NEG | N | Negative charge |
| ACID | C | Acid |
| BASE | B | Base |
| AROM | R | Aromatic ring |
| LIP | L | Lipophile |
| MET | M | Metal atom |
| LPD | X | Lone-pair donor (Lewis base) |
| USR1 | W | User-defined centre type 1 |
| USR4 | Z | User-defined centre type 4 |
The user may elect to restrict the pharmacophores to a subset of these centre types. The centre type for each atom is defined by its atom type. The default atom types do not include sulphur as a hydrogen bond acceptor but does include it as a hydrogen bond donor.
Fuzzy pharmacophores are designed to handle the tolerances on the exact positions of the centres (atoms) that define a pharmacophore, which affects the distances between the atoms. When calculating a pharmacophore, THINK allows a tolerance of ±x on each distance. This has the effect of "spreading" the distance across several distance bins, leading to the creation of multiple pharmacophores for a single set of centres. Each of these pharmacophores can be considered to be a fraction of the original pharmacophore. The pharmacophore count is adjusted to take into account these fractional pharmacophores, so that the total count remains unchanged.
For example, a 2-centre pharmacophore has a distance of 4.85Å between the centres. Applying a pharmacophore tolerance of 0.25Å gives a range of possible distances between 4.6 and 5.1Å. The default distance bins that cover this range are 4.0-5.0Å and 5.0-6.0Å: 80% of the distance range lies within the 4.0-5.0Å bin and 20% within the 5.0-6.0Å bin. Thus, this 2-centre pharmacophore would generate two fractional pharmacophores, one with a count of 0.8 and the other with a count of 0.2.
Since the pharmacophore tolerance is applied independently to each of the distances within a pharmacophore, the number of fractional pharmacophores created increases dramatically as the number of centres, and hence the number of distances, rises. This means that pharmacophore calculations may run more slowly than in previous versions, although the algorithms have been speeded up in compensation.
The pharmacophore tolerance x is taken from the 3D tolerance stored in the common variable D3DTOL so that 2x = D3DTOL. The tolerance may be changed through the command CUSTOMISE TOLERANCE=value. Results from earlier versions of THINK can be reproduced by setting the 3D tolerance to zero.
7.3 Pharmacophore profiles
A pharmacophore profile is the set of all pharmacophores found across the conformers of a series of molecules. As each individual pharmacophore is generated during the calculation, it is:
Note that the pharmacophore profile shows the spread of pharmacophores across the series of molecules. It does not calculate the sum of the pharmacophores exhibited nor is it normalised. A pharmacophore profile may be saved to a file.
| SAVE FORMAT=PROFILE FILE=file |
| SAVE FILE=filename.pro |
7.4 Pharmacophore Files
Pharmacophores are normally saved to a file for analysis by another software program. To facilitate the movement of these files between different operating systems a formatted file is used, with the pharmacophore data written in a compressed format using 6 (3-centre) or 10 (4-centre) characters to describe each pharmacophore. If conformer-based counts are enabled then the number of times each pharmacophore occurred will also be saved, increasing the number of characters used per pharmacophore. A full description of the format can be found in Appendix B.
8 Molecule Searching
THINK includes functionality to search molecules stored in a SMILES or MDL SD file for a 2D substructure, a 3D pharmacophore and by similarity (based on functional groups). The results of the search are normally stored in a SMILES file for 2D searches and in an SDF file for 3D searches. It can sometimes be helpful to consider the exact match search to be a special case of substructure searching and the R-group search to be a special case of substructure searching. The site search is a variant of 3D searching. Using the display features of THINK, the hits can be viewed with the query superimposed for 2D searches, or superimposed on the pharmacophore for 3D searches. When queries are read from files, the option to omit implicit hydrogens can be essential.
The substructure searching allows a query consisting of a molecular fragment to be found in a subset of the atoms within the molecule being searched. In the exact match search, all the atoms in the molecule being searched must be matched; this means that the exact match search can be used to check for duplicates. More advanced substructure searching options use atom wildcards consisting of reserved atom types from the following list:
| Atom type | Query |
| A | Any atom |
| A- | Any atom with formal charge of -1 |
| A2- | Any atom with formal charge of 2 |
| A+ | Any atom with formal charge of +1 |
| A2+ | Any atom with formal charge of +2 |
| A3+ | Any atom with formal charge of +3 |
| M | Metal atom |
| R | Carbon or hydrogen |
| X | Oxygen or nitrogen |
| Q | Any atom except hydrogen or carbon |
| Z | Any atom except hydrogen |
| HAL | Any halogen |
| CAK | Chain carbon |
| CRG | Ring carbon |
| CAL | Non-aromatic carbon |
| CAR | Aromatic carbon |
| NAK | Chain nitrogen |
| NRG | Ring nitrogen |
| NAL | Non-aromatic nitrogen |
| NAR | Aromatic nitrogen |
| OAK | Chain oxygen |
| ORG | Ring oxygen |
| OAL | Non-aromatic oxygen |
| OAR | Aromatic oxygen |
| SAK | Chain sulphur |
| SRG | Ring sulphur |
| SAL | Non-aromatic sulphur |
| SAR | Aromatic sulphur |
| GP01 | Any element from periodic table group 1 |
| GP02 | Any element from periodic table group 2 |
| GP03 | Any element from periodic table group 3 |
| GP04 | Any element from periodic table group 4 |
| GP05 | Any element from periodic table group 5 |
| GP06 | Any element from periodic table group 6 |
| GP07 | Any element from periodic table group 7 |
| GP08 | Any element from periodic table group 8 |
| GP09 | Any first row transition element |
| GP10 | Any second row transition element |
| GP11 | Any third row transition element |
| GP12 | Any lanthanide |
| GP13 | Any actinide |
The similarity search uses a functional group fingerprint where each bit is set when the corresponding functional group is present in the molecule. The list of functional groups is defined using SMILES and stored in the file key.smi (stored in the THINK_EXEC directory). A Tanimoto similarity index is derived from the functional group fingerprints of the query and the molecule being searched according to the following formula:
![]()
where NC is the number of common bits set in the query and the molecule; NQ is the number of bits set in the query and NM is the number of bits set in the molecule.
Molecules are accepted as hits when the similarity index exceeds the specified cutoff.
This is a pre-requisite step to generate files containing R-groups prior to enumerating libraries, unless the user wishes to define all the R-groups manually. The implementation in THINK v1.25 is appropriate to generate R-groups for core-based and coreless libraries but cannot generate peptide building blocks with two generic connections. (This would require the SMILES to be written in an atom order matching that of the query). When enumerating a coreless library, THINK uses one group in place of a core. As a result, this section only considers core-based libraries but is equally applicable to coreless libraries. For most libraries, the choice of the core group has implications for the allowed R-groups. Occasionally, it is necessary to perform a substructure search as a preliminary step to identify certain R-groups.
The most convenient query is usually a reaction with reactants including substitution positions and product(s) which form the core group for enumeration.
 |
[1]C(=O)Cl+[2]N(H)H>[1]C(=O)N(H)[2] |
Each reactant is an R-group query consisting of a substructure with the desired connection points. During the search, atoms in the reagent that are matched to the substructure are deleted, and atoms matched to the connection points are replaced with generic or explicit connection atoms (see section 2.4). THINK v1.25 does not support graphical entry of the query and consequently it is usually created as a SMILES string, with the connection points indicated by:
For instance, a query to search for reagents of the form [R1](Cl)C=O, where [R1] is the required R-group, would have the form [0](Cl)C=O if the R-group is to have a generic connection atom (so the R-group can be used in any position) or [n](Cl)C=O for an explicit connection atom (which restricts the R-group to a single position).
Each connection point in the query is treated as a wildcard atom type during the search. The Z wildcard will match any atom type except hydrogen (this prevents the R-group search creating R-groups consisting of single hydrogen atoms). Use of the [wildcard] connection allows a more powerful search to be performed by restricting the connection point to a subset of atom types
If the query contains multiple connection points, it is recommended that they are specified as explicit connections using [n]. Where this is not possible (for instance if the [wildcard] form is required), the connection numbers may be defined by setting the group numbers of the connection atoms. Once the query has been read into THINK, the group number of an atom can be changed using the MODIFY CHANGE=atom GROUP=n command. If a search is performed using a query that contains two or more generic connections, THINK will detect the situation and use explicit connection atoms in the resulting R-groups. However, the order in which these connections are allocated is undefined, so the search may not give the desired results. It is therefore recommended that explicit connections are always used in these circumstances.
The search eliminates duplicate R-groups by omitting the second and subsequent copies of the R-group from the results file. In THINK v1.25 molecules with more than one occurrence of the substructure are skipped.
The 3D search has been designed principally for pharmacophore searching. In THINK the query consists of the substructure (such as there may be) of the specified molecule plus the inter-centre distances.
The 3D search starts by performing a 2D search to find the matching 2D substructure within the molecule being searched. Up to 1000 separate occurrences of the substructure will be processed; each is known as a substructure solution. A path test is used to eliminate substructure solutions that cannot possibly meet the query. This considers the connected path or chain between atoms in the molecule which correspond to atoms in the query that have a distance constraint. The maximum possible distance is calculated for this chain (ie if the chain were fully extended). The substructure solution is discarded when this distance is less that the minimum required by the distance constraint. For 1,2, 1,3 and 1,4 connection paths, the minimum possible distance for the chain is also estimated and used to eliminate substructure solutions.
Every distance check in a 3D search includes a small tolerance. The tolerance used for each check is the larger of the two following values: the CUSTOMISE TOLERANCE setting, or the sum of the radii of the two query atoms defining the distance (when this is enabled using the CUSTOMISE RADIUS=TOLERANCE command).
A conformational analysis is then performed using the current settings (see Chapter 4). A conformer which meets the geometry requirements is fitted to the query (using the usual Ferro Hermans fitting algorithm, preceded by an approximate match based on best-plane projections) and saved. The serial numbers of the conformer are modified to match the query or set to >512 for those atoms which do not correspond to any in the query.
The conformational analysis stage of a 3D search continues until all conformers have been scanned, checking all possible substructure solutions for each conformer. A new hit is created for each acceptable substructure solution for each conformer. If the results are being written to an SD file, all hits are saved in the file. If the results file uses the SMILES format, the best hit (the one that most closely matches the query) will be saved in the file. The maximum number of hits created for each conformer is controlled by the common variable ICFHMX, and the maximum number of hits for each molecule by the common variable IMLHMX. Both variables are normally set to zero, meaning that no limit is imposed.
Prior to performing the conformational analysis, the bonds being rotated are ordered so that bonds which do not change inter-atomic distances that match those in the query are incremented first, an d bonds for ring conformational analysis are changed last. This accelerates the conformational analysis because bond rotations that do not change critical inter-atomic distances may be skipped until a conformer is found which matches the query. Ring conformational changes are most computationally intensive, and by placing these at the end of the list they are repeated the minimum number of times.
When the search results file is viewed in THINK with the query, the conformers are not automatically refitted to the query with the consequence that rotations and translations of the query will ruin subsequent viewing.
The site search is a useful technique for virtual screening in order to select molecules that have a high probability of interacting with a protein. For a given protein receptor site, all the ways in which molecules might interact are not usually obvious. The query consists of a set of pharmacophore centres which complement those in the receptor and represent the ideal interactions with the receptor (see chapter 9 for information on creating a site query). These centres may consist of any mixture of the 10 standard (HDON, HACC, POS, NEG, ACID, BASE, AROM, LIP, MET, LPD) and 2 user-defined (USR1, USR4) centre types, and form the query for a site search. A molecule is required to fit any pharmacophore defined by 2, 3 or 4 of these centres (depending on the CUSTOMISE settings), whereas in a normal 3D search the molecule would be required to fit all the pharmacophores defined by the centres. In addition, the pharmacophore must exhibit the minimum pharmacophore score (see section 9.5.2) set by CUSTOMISE WARP=value and each centre must originate from different residues if CUSTOMISE RESIDUE=DIFFERENT has been issued.
In normal usage it would also be appropriate to have the active site residues present in a separate molecule in order to score the solutions (and eliminate those containing atoms which collide with the receptor). The receptor site residues are normally read from a PDB file and the complementary centres may either be in a PDB or SDF file. PLUS centres are automatically renamed to POS centres as they are read into THINK. The serial numbers of the centres usually map to receptor site atoms with which they interact (to aid visualisation), and this can require use of an extension to SD files in order to support serial numbers exceeding 999 (see section 2.2.2).
In an analogous manner to 2D and 3D searches (see sections 8.1 and 8.4), for each molecule the program first constructs a list of atoms that may match each centre. Permutations of 2, 3 or 4 centres are then constructed and the connected paths between these atoms evaluated to determine whether the observed separation (within the allowed tolerances) might be achievable in a conformational search. As in 3D searches, a tolerance is applied to each distance check. This generates the list of substructure solutions.
In addition, the area of the 3-centre pharmacophore or volume of the 4-centre pharmacophore is compared with the number of non-hydrogens atoms in the molecule. Where the volume (or area) is less than the product of the number of non- hydrogens atoms and the CUSTOMISE VOLUME (or CUSTOMISE AREA) setting the permutation is ignored. The conformational analysis is performed only if there are some permutations that might meet the distance and volume (or area) criteria.
For conformers which match a pharmacophore, the conformer is fitted to the receptor using the matched pharmacophore. For 2-centre pharmacophores, an additional pair of points is required to fit the conformer. These are generated from the centroid of the molecule and the centroid of the complementary centres defining the pharmacophore. The fitted conformer is scored using an extended ChemScore function:
![]()
where:
DG0,
DGhbond, DGlipo,
DGrot and DGbad
are constants (-5.48; -3-34; -0.117; 2.56; 0)
Nhbond is the number of qualifying interactions (on geometric
criteria)
Nlipo is the number of lipophilic-lipophilic contacts
Nrot is the number of frozen rotatable bonds in the molecule
Nbad is the number of lipophilic-hydrophilic contacts
and
EVdW is the VdW interaction energy between the molecule and protein, computed by the expression:
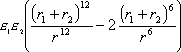
where:
e1
and e2 are constants
for the atoms
r1 and r2 are the VdW radii for the atoms
r is the distance between the atoms
and
Etors is the torsional energy of the molecule, computed by the expression:
![]()
where:
Vsng and Vcnj are constants (1.35; 3.72)
q is the torsion angle
Conformers that score less than the initial cutoff (common variable GSTMIN default 1000) are then refined using a Simplex minimiser to improve the estimated free energy of binding. The minimiser will adjust the position, orientation and exact conformation of the ligand within the active site. If the resulting conformer score is below the search cutoff value (common variable GSTHIT), the conformer is accepted as a hit. The search proceeds with the next substructure solution or continues the conformational analysis after finding a hit conformer. At the end of the conformational analysis the best hit (based on score) is saved to the results file. If all acceptable hits are required, the common variable ISTALL should be set to one, and the results file must use a 3D format (eg SD file).
Use of the Simplex minimiser to refine each hit conformer is important when attempting to reproduce crystal ligand-protein binding geometry. It is sometimes necessary to decrease the torsional increment by increasing the number of points about one or more classes of rotatable bonds (see section 4.1). It is conceivable that the optimised ligand will no longer fit the pharmacophore (within the tolerances) unless it is constrained to do so. This was suggested by C A Baxter et al. In THINK, when the deviation of the matched atoms exceeds the tolerance there is an additional contribution to the free energy consisting of K(d-t)2, where K is the value of the common variable GSTRES (default 1000), d is the deviation and t is the tolerance. Setting GSTRES to 0 disables the constraint.
8.6 Checkpoint File and Search Tracing
If a search is interrupted due to an unexpected halt in the application (eg a power failure), it can be resumed from the point at which it was interrupted. This is done by means of a checkpoint file called "search.dat" which is written to the current working directory during each search. The file stores information on the progress of the search together with details of query being used, the file being searched and the name of the results file. If the search is restarted in a subsequent THINK session using the same query and file names, the first part of the search will be skipped and progress will resume from the data stored in the checkpoint file. If the whole search is to be repeated, it is important that any existing checkpoint file is first deleted.
The checkpoint file will be deleted if a search is manually interrupted through <CTRL-C> or the Cancel button on the progress report.
It is possible to record the destiny of the molecules being searched to the THINK console window and log file. When searching a large number of molecules, this additional output can be voluminous and time-consuming. Consequently, the option should only be used when attempting to analyse why a molecule was not found, and it is recommeded that the number of molecules being searched is reduced to the smallest possible subset. The additional output is enabled through the common variable ITRACE - this is a bit mask, and setting bit 0 (value 1, in other words setting ITRACE to an odd number) will enable the extra output.
The approach used to generate a site query for a protein depends on whether the coordinates of a ligand docked into the active site are available. If they are not then it is essential to have a list of the residues which constitute the active site (THINK does have functionality to search for potential binding sites).
THINK will identify the key atoms within the active site and insert the appropriate complementary centres, also known as site points, after checking atom overlap and accessibility as described below. The centres are placed in a new molecule called INTERACT. It is recommended that this molecule is saved in a PDB file for re-use in later searches.
The following table shows the atom types of the key atoms and their corresponding complementary centres (see section 7.1 for a description of the codes used to describe the centres). The position codes (A,B,C) identify where the centres will be inserted, and are described after the table. Each centre is allocated a VdW radius of 1.0Å, which is used to determine the tolerance used in the distance checks during site searches (see section 8.5).
| Atom type |
No. of non-H
connections |
Tautomeric?
|
Complementary centres |
Position
code |
| OSP2 |
1
|
N
|
HDON, HACC |
A
|
|
Y
|
HDON, HACC, POS |
B
|
||
|
2
|
N
|
HDON |
A
|
|
| O- |
1
|
N
|
HDON, POS |
A
|
| OAR |
2
|
N
|
HDON |
A
|
| OSP |
1
|
N
|
HDON, POS |
B
|
|
Y
|
HDON, HACC, POS |
B
|
||
| NSP2 |
1
|
N
|
HACC |
B
|
|
Y
|
HDON, HACC |
B
|
||
|
2
|
N
|
HACC |
A
|
|
|
Y
|
HDON, HACC |
A
|
||
| ND |
1
|
Y/N
|
HDON, HACC |
B
|
|
2
|
N
|
HDON |
A
|
|
|
Y
|
HDON, HACC |
A
|
||
| N+ |
1
|
Y/N
|
HACC, NEG |
A
|
|
2
|
Y/N
|
HACC, NEG |
A
|
|
|
3
|
Y/N
|
HACC, NEG |
A
|
|
| ND+ |
1
|
Y/N
|
HACC |
B
|
|
2
|
Y/N
|
HACC |
A
|
|
| NSP3 |
1
|
Y/N
|
HDON, HACC, NEG |
A
|
|
2
|
Y/N
|
HDON, HACC, NEG |
A
|
|
|
3
|
Y/N
|
HDON, HACC, NEG |
A
|
|
|
4
|
Y/N
|
NEG |
A
|
|
| NAR2 |
2
|
N
|
HDON |
A
|
|
Y
|
HDON, HACC |
A
|
||
| NAR3 |
2
|
N
|
HACC |
A
|
|
Y
|
HDON, HACC |
A
|
||
| AR5 |
-
|
-
|
AROM |
C
|
| AR6 |
-
|
-
|
AROM |
C
|
With position code A, the complementary centres are inserted at a single position 3.0Å from the defining protein atom projected along a vector defined by the sum of the non-hydrogen connections to that atom.
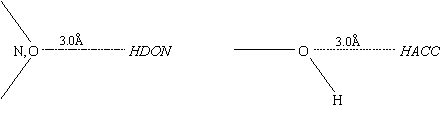
Position code B inserts two sets of complementary centres. The two insertion points and the defining atom form an equilateral triangle with the centre at the apex. The sides of the triangle are 3.0Å and the base (between the two insertion points) is 2.0Å. The midpoint of the base is positioned on the vector projected along the non-hydrogen connection to the defining atom.
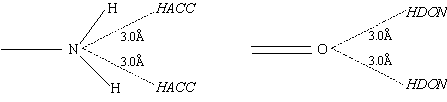
Position code C is used with aromatic rings and inserts two centres 3.2Å above and below the plane of the ring (positioned directly above/below the defining AR5 or AR6 atom).
THINK creates several complementary centres at a single insertion point because the defining atom often has more than one centre attribute. For instance, O- is both a hydrogen bond acceptor and a negative charge centre; consequently it requires two complementary centres (HACC and POS).
Insertion points which are inside the VdW radius of any atom in the protein molecule are eliminated. This can be adjusted by modifying the common variable RADCEN (which is zero by default). A negative value permits some overlap with the protein while a positive value might allow for the absence of hydrogens.
An accessibility check is applied to each insertion point by examining the vector from the defining protein atom to the insertion point (the insertion vector). If the vector points away from the centre of the active site, the defining atom is unlikely to be accessible by a ligand and therefore its complementary centres are not created. The check is actually performed by determining the angle between the insertion vector and the vector from the centroid of the active site to the protein atom. If this angle is less than 60° the insertion point is rejected. The angle can be changed by storing the cosine of the minimum permitted angle in the common variable COZCEN.
9.2 Existing Ligand-Protein Complex
The most common existing ligand-protein complexes used in validation are X-ray crystal structures, although interactively docked ligands and ligands docked using other software may also be used. THINK can deduce the active site from the position of the ligand, so this technique can be used with PDB files that do not contain a definition of the site.
If the ligand and the protein exist in the same PDB file it may be necessary to edit the PDB file and insert records containing the NAME keyword to allocate names to the molecules and indicate where additional molecules start. In some PDB files, where the ligand is not a peptide, THINK will automatically recognise the separate molecules providing the appropriate HETNAM records are present. When editing PDB files, it is important to move any HETATM and CONECT records to the appropriate place to be associated with the correct molecules defined by the NAME records. If there are connections between the protein and ligand, these must be removed by editing the file.
It is recommended that all non-essential water molecules are deleted before the site query is generated. In some cases, however, the ligand may bind to the protein via one or more essential water molecules. If the user decides to retain these essential molecules, the remaining water molecules must be removed by editing the PDB file (remembering to move any CONECT records into the right place).
In order to create the complementary centres or site points, THINK first needs to define the active site by identifying residues that contain at least one atom in contact with the ligand. In most cases it is appropriate to use a contact radius which is larger than the VdW radius by about 25% to allow residues that are close to, but not in VdW contact with, the ligand to be included in the site. The ratio of the contact radius to the VdW radius is specified as part of the command to create the complementary centres.
| MODIFY INTERACTION=^1ABC LIGAND=ligand |
THINK then creates complementary centres for all key atoms within the residues in the active site, subject to the overlap and accessibility checks described in section 9.1 and places them in a new molecule called INTERACT. For large sites, this can give a large number of centres which significantly increases the time required for a site search. However, the presence of a ligand allows THINK to allocate a weighting to each centre reflecting its distance from the ligand, and hence the likelihood that its originating protein atom is involved in the ligand binding. The weights are stored as group numbers for the centres, and are described in section 9.5.3.
9.3 List of Active Site Residues
If the PDB file does not contain a bound ligand, the set of residues in the active site must be supplied when a site query is created. The set may be defined by:
The comma-separated list must not contain any spaces
9.3.1 Site Records in PDB file
When the active site is defined by SITE records in the PDB file, THINK will interpret these records and store the definition as a list of residues using symbols of the form SITE-name(n) where name is the PDB identifier for the SITE and n is a counter for the list of residues. If this information is missing from the PDB file, it can be specified using commands. For example, to create a site with an X identifier defined by three residues with sequence numbers 905, 982 and 909, then the following commands would be used:
| LET SITE-X(1) := (905) LET SITE-X(2) := (982) LET SITE-X(3) := (909) LET SITE-X(4) := " " MODIFY INTERACT=_SITE-X |
The blank stored in SITE-X(4) terminates the list of residues. It is essential if the SITE-X symbol is being re-used with a new definition - if it was omitted, the commands would be interpreted as re-defining only the first three elements of SITE-X, not the whole symbol. If these commands are issued in the console window then it is not necessary to specify global symbols (using ":=") rather than local ones. However, if the definition is stored in a logfile global symbols must be used.
9.3.2 Finding Binding Sites
When the binding site is unknown, THINK can be used to find all plausible binding sites and stores the results in a symbolic array of the form SITE-x(n) where x is the site number and n is a counter for each residue in the site. The following algorithm is used:
The following command is used:
| MODIFY SITE=protein |
After the INTERACT molecule containing the site points has been created, THINK modifies the protein by eliminating residues which are so far from the active site that they cannot make any contribution. Reducing the protein to an "extended site" speeds up the search times, and is essential when THINK is used in a distributed computing environment because there is less data to transfer between the servers and the clients. Therefore, it is recommended that the extended site is saved in a PDB file for re-use.
An extended site comprises all residues that contain at least one atom within a given distance d of any complementary centre in the INTERACT molecule. The distance d is defined as (8.0+Ra+Rc) where Ra and Rc are the VdW radii of the atom and centre respectively. The value of the additional distance, 8.0Å, is stored in the common variable RADSIT and may be altered if desired.
The following command would create the site points for a protein called 821P with a bound ligand in a molecule called BOUND. The resulting query molecule (INTERACT) and extended site are saved in PDB files so they can be re-used in multiple searches.
| MODIFY INTERACT=^821P LIGAND=BOUND RATIO=1.25 FILE SAVE=821P-Q1.PDB MOLECULE=INTERACT FILE SAVE=821P-SITE.PDB MOLECULE=821P |
9.5 Centre Selection (Weighting)
The choice of pharmacophores to be used in the search is influenced by the weightings of the complementary centres (site points). There are three different types of weightings:
These weightings are combined to select the best pharmacophores from all possible permutations of the site points during the search.
The weightings are stored as group numbers for the complementary centres, and may be set automatically by THINK and/or manually by the user. The group numbers are also stored in PDB files (as an extension to the standard format - see section 2.3.2). They are often useful when manually editing the query file to reduce the number of centres, as they can be used to guide the choice of centres to be removed.
Once the complementary centres have been created, they can be reviewed to locate the points most likely to be involved in the ligand binding. These may be grouped to create a list of alternatives for each point in a pharmacophore. This can be important when molecules are required to interact with one or more residues. When THINK constructs the pharmacophores from the site query, it will check for these lists of alternatives and construct the pharmacophores appropriately.
Up to four sets of complementary centres may be defined, identified by their group numbers. The group number is used as a bit mask, which means that the sets may overlap by using group numbers that correspond to two or more bits being set (adding the individual group numbers together).
| Required group | Bit | Group number |
| 1 | 4 | 16 |
| 2 | 5 | 32 |
| 3 | 6 | 64 |
| 4 | 7 | 128 |
For instance, a centre with group number 16 would be included in required group 1 only, whereas a centre with group number 48 (16+32) would be included in required groups 1 and 2.
THINK automatically constructs the lists of alternative pharmacophore points by scanning the group numbers of all complementary centres. All centres that do not fall into one of the above groups are placed into group 0. This group is used to provide pharmacophore points that cannot be selected from one of the user-defined groups. THINK ensures that all the centres used in the pharmacophore have been derived from different atoms in the protein or ligand by checking their serial numbers.
Once the pharmacophores have been constructed (taking the required groups into account) THINK allocates each an initial score of 100. This score is reduced by the distance, acid and base weightings to give the final score for each pharmacophore. Only pharmacophores with scores greater than the common variable PSCOMN will be used during the search. This value is changed using the CUSTOMISE WARP command.
If the site query is derived from an existing ligand-protein complex, THINK automatically allocates a distance-based weighting to each complementary centre generated. This reflects the distance of the centre from the ligand, and hence the likelihood that its originating protein atom is involved in the ligand binding. These weights are also stored as group numbers and can be used in conjunction with the required groups described above by adding the group numbers together.
| Group number | Interpretation |
|
1
|
Centre within 1.0Å of a ligand atom |
|
2
|
Centre in contact with ligand atom (separation is less than 1.0+Ra where Ra is the VdW radius of the ligand atom) |
|
3
|
Centre is within 6.0Å of a non-hydrogen ligand atom and there are no intervening non-hydrogen protein atoms between the centre and 3 or more non-hydrogen atoms in the ligand (ie "line of sight") |
|
4
|
Centre is more than 6.0Å from all non-hydrogen ligand atoms, or there is no line of sight to the ligand |
|
5
|
Centre has fewer than 5 residues infront of it (based on the plane perpendicular to the vector from the centre to the originating atom). |
When a query is created from a binding site weightings are still applied although these critically depend on the centroid of the residues defining the site.
| Group number | Interpretation |
|
1
|
Site point within 6.0Å of site centroid |
|
2
|
Site point within 8.0Å of site centroid |
|
3
|
There are no intervening non-hydrogen protein atoms between the site point and the site centroid (ie "line of sight") |
|
4
|
There is no line of sight to the site centroid |
|
5
|
Centre has fewer than 5 residues infront of it (based on the plane perpendicular to the vector from the centre to the originating atom). |
For instance, a site point may be allocated a group number of 19, which means that it is included in required group 1 (value 16) and has a distance weighting of 3.
When a centre with a distance weighting is used in a pharmacophore, the pharmacophore's score is divided by the distance weighting (group number). For example, a pharmacophore containing four centres with distance weightings of 1, 2, 2 and 3, the score for that pharmacophore would be 8.3 (100 divided by 1*2*2*3). The pharmacophore score is important because those scoring below PSCOMN are ignored.
The acid and base weightings are used in conjunction with the distance weightings to accept only acidic and basic groups that lie very close to similar groups in the original ligand, and therefore are likely to interact in the same way.
Most acidic groups contain a hydrogen donor, so an acid group in a potential ligand may be matched to an ACID or HDON site point. Similarly, basic groups normally contain a hydrogen acceptor, so may be matched to a BASE or HACC site point. THINK prefers to match an acid group to an ACID point, and a basic group to a BASIC point, so penalises pharmacophores where the groups are matched to HDON or HACC points by reducing the pharmacophore score.
The penalty for acid groups is stored in the common variable PSACID, and for basic groups in the variable PSBASE. Both variables contain a value in the range 0-1. For each HDON point in the pharmacophore matched to an acidic group, the pharmacophore score is multipled by the value in PSACID. Similarly, for each HACC point matched to a basic group the score is multipled by the value in PSBASE.
9.6 Reproducing Ligand-Protein Complexes
Attempting to reproduce a known ligand-protein complex can be a useful way of validating the results generated by THINK. In some cases creating the site query using the default options and then running the site search using the ligand as the search molecule reproduces the geometry of the original ligand, but this is not always the case. For instance, some ligands may bind using only weak interactions (no hydrogen bonds or charge interactions), or may bind through a water molecule that was missed when the site query was created. It is possible that the crystallographic data shows the mean position of the ligand, where there are no strong interactions, and that the ligand may move between positions where hydrogen bonds etc may be formed. THINK also takes no account of the flexibility of the side chains within the active site.
THINK can identify the key interactions between the ligand and the protein using the following command:
| DISPLAY MODE=3D MOLECULE=protein QUERY=ligand OPTIONS=INTERACTIONS |
which lists the interactions to tbe console window and draws dashed lines between atoms to represent the interactions. The interactions are a subset of the contacts between the ligand and the protein: only contacts between atoms that are complementary pairs of interaction centres are included:
For proteins without hydrogen atoms, THINK will detect hydrogen bond donor atoms despite the absence of the hydrogens (normally a hydrogen bond donor requires at least one attached hydrogen atom unless the CUSTOMISE DONOR=NOHYDROGEN command has been issued). Any water molecules present in the protein will be included in the list of interactions, and THINK will show hydrogen-bonding pathway through water molecules (see section 2.3.3 for details of how water molecules are handled when a PDB file is read). If a search fails to reproduce a known complex, it may be worth re-examining the site with the water molecules included in the protein in case any are involved in the binding.
When determining the contacts, THINK uses an enlarged VdW radius for each atom. By default, a factor of 1.25 is applied to each radius. However, if "super-CPK" radii are in effect, these will be used instead (super-CPK radii are enabled by setting the ratio of the CPK to VdW radius to a value greater than 1.25 using the CUSTOMISE RATIO keyword).
9.6.2 Check Site Points
Comparing the complementary centres (site points) in the query with the set of interactions reported from the ligand-protein complex may reveal some interactions that do not have corresponding site points. These points were probably eliminated when the query was created because:
The rules controlling the insertion of site points are described in section 9.1. Changing the common variable RADCEN to a negative value will allow THINK to create site points inside the protein.
The accessibility check may not be appropriate for a large or irregularly shaped active site, in which case it may be relaxed by setting the common variable COZCEN to a value closer to 1.0 (the default value is 0.5). The check can be disabled by setting COZCEN to zero.
9.6.3 Check Pharmacophores and Conformers
It is not uncommon for a ligand to have only two or three points that interact with the protein. For these examples the number of centres in the pharmacophores must be reduced from the default value of four. Use of a 3- or 4-centre pharmacophore is normally sufficient to force the ligand into the desired conformation, but a 2-centre pharmacophore often leaves scope for much of the molecule to adopt a different conformation. Consequently, comparing the site search hits with the docked ligand may not be encouraging.
The results of a site search may be analysed in the spreadsheet. To obtain the maximum information, the protein, the original bound ligand (obtained from the crystallographic data) and the 3D coordinates of the search hits must be read into THINK before the spreadsheet is calculated. This means that the search results must be stored in an SD file.
The spreadsheet is generated by the command:
| LIST INFO=PROPERTIES OUTPUT=WINDOW QUERY=ligand |
It may be necessary to restrict the properties calculation to the hit molecules by adding the keyword "MOLECULE=hits" to the command. THINK automatically computes the following RMS deviations of each molecule from the ligand (as specified with the QUERY keyword) and stores the values in fields within the spreadsheet:
| Field name | Description |
| RMS-All | Deviation of all atoms, based on atom order within the molecules - only computed if both molecules have the same number of atoms |
| RMS-NoH | Deviation of all non-hydrogen atoms in hit from corresponding atom (based on atom order) in ligand - only computed if both molecules have the same number of atoms |
| RMS-Match | Deviation of all atoms with matching serial numbers (excluding those in group 999) |
The RMS-ALL and RMS-NOH fields are intended for use in validation studies when the original ligand (as taken from the crystallographic data) is used as the molecule being searched. These fields show the deviation of the hit conformers from the original conformation.
The RMS-MATCH field can be used with any hit molecule found by a search using a site query derived from the original ligand, regardless of the number of atoms in the molecule. It uses the serial numbers of the atoms to establish their correspondence (the serial number of an atom in the hit molecule is taken from the matching atom in the query, which is itself copied from the defining atom in the original ligand). Atoms in group 999 are omitted as they were not matched to an atom in the site query.
There is a fourth RMS field in the spreadsheet, called "RMS". This shows the RMS deviation of the matched atoms in the hit from the corresponding atoms in the query (not the original ligand). Its value is computed during the site search and is saved in the search results file.
If the active site is present when the spreadsheet is computed, THINK will re-calculate the docking score for each hit molecule (see section 8.5) and include it, together with a breakdown of the score, in the following fields:
| Field name | Description |
| G-TOT | Total docking score |
| G-HB | Hydrogen-bonding component (DGhbond*Nhbond) |
| G-LIP | Lipophilic contacts (DGlipo*Nlipo) |
| G-ROT | Frozen rotatable bonds (DGrot*Nrot) |
| G-NB | Non-bonded component (EVdW) |
| G-BAD | Lipophilic-hydrophilic contacts (DGbad*Nbad) |
It may be necessary to explore various search options using trial and error in an attempt to understand why THINK is failing to dock a known ligand. The time taken to do this can be reduced by eliminating site points that do not interact with the protein. Such points can be identified by their group number: those in group 4 are too far away from the protein to contribute to any interaction. It is also recommended that the trace option is enabled (by setting the common variable ITRACE) during the trials.
| LET #ITRACE = 1025 |
Setting ITRACE to 1025 will provide additional output which usually indicates at what stage in the search the molecule is eliminated (see Appendix D). If the explanation of why the search failed is "Atoms not matched", it implies one of the following:
In other cases, the explanation is "no suitable conformers" which is indicative of either the torsional increments being too large or the conformers being eliminated due to contacts either within the molecule or between the molecule and the protein. It is not unusual to reduce the ratio of the CPK to VdW radius from 0.6 to 0.4 to remove some of the contacts. For crowded conjugated systems, the torsional sampling about conjugated bonds may be increased to 4.
In the event of a conformer meeting the geometry constraints but failing to generate an adequate score adequately, further explanations will be provided. Significant numbers of bad contacts between the ligand and the protein may lead to the explanation "score too high to refine". In such cases, it is possible to increase the maximum number of cycles of minimisation by increasing NCYCMX. With 3- and 4-centre pharmacophores, it may be preferable to increase the number of torsion increments to provide a better starting geometry rather than increasing GSTMIN above the default of 1000. (GSTMIN is the maximum permitted score for a molecule to enter the refinement stage: increasing this value will lead to more molecules being accepted for refinement, thereby increasing the overall search time). Using a SEARCH CUTOFF value that is equal to GSTMIN will eliminate the refinement step, which can be useful to provide output for visually checking that the appropriate solutions would otherwise be generated.
Appendix A Parameter File Formats
The initial letter of all variable names in the following descriptions indicates the type of variable:
Blank records and records beginning with ! are skipped when the file is read.
A.1 Atom Types
| Data: | CELEM, CTYPE, IVALEN, INUMB, ICOLOR, IATTRIB, IFORMQ, IGROUP, ICONN, RRAD, RWEIGT, RENEG, REVDW | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Format: | A4, 1X, A4, I3, I4, I3, I9, 3I3, F6.2, F8.2, F5.1, F7.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CELEM | A4 | Element symbol | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTYPE | A4 | Atom type | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IVALEN | I3 | Valency | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| INUMB | I4 | Atomic number | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ICOLOR | I3 |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IATTRIB | I9 |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IFORMQ | I3 | Formal charge | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IGROUP | I3 | Group or row in periodic table (1-13), corresponding to the numbers used for the 2D search wildcard atom types (see section 8.1) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ICONN | I3 | Number of connected atoms | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RRAD | F6.2 | VdW radius | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RWEIGT | F8.2 | Atomic weight | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RENEG | F5.1 | Electronegativity | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| REVDW | F7.3 | VdW e term (see section 8.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A.2 Bond Parameters
| Data: | CTYPE1, CTYPE2, IORDER, RLEN, RCONST | ||||||||||||||||||||||
| Format: | A4, 1X, A4, I3, F7.3, F9.2 | ||||||||||||||||||||||
| Description: | |||||||||||||||||||||||
| CTYPE1 | A4 | Atom type for first atom defining bond | |||||||||||||||||||||
| CTYPE2 | A4 | Atom type for second atom defining bond | |||||||||||||||||||||
| IORDER | I3 |
|
|||||||||||||||||||||
| RLEN | F7.3 | Ideal bond length | |||||||||||||||||||||
| RCONST | F9.2 | Force constant | |||||||||||||||||||||
A.3 Angle Parameters
| Data: | CTYPE, RANGLE, RCONST, RCOS | ||
| Format: | A4, 2F7.1, F9.4 | ||
| Description: | |||
| CTYPE | A4 | Atom type | |
| RANGLE | F7.1 | Ideal bond angle (in degrees) | |
| RCONST | F7.1 | Force constant | |
| RCOS | F9.4 | Cosine of RANGLE | |
A.4 Ring Coordinates
| Data: | ISIZE, IORDER, CELEM, X, Y, Z | ||||||||||||||||||||||
| Format: | 2I3, 1X, A4, 3F8.3 | ||||||||||||||||||||||
| Description: | |||||||||||||||||||||||
| ISIZE | I3 | Number of atoms in ring, or 0 for next ISIZE-1 records | |||||||||||||||||||||
| IORDER | I3 |
|
|||||||||||||||||||||
| CELEM | A4 | Element for atom n | |||||||||||||||||||||
| X, Y, Z | 3F8.3 | XYZ coordinates | |||||||||||||||||||||
A.5 Ring Torsion Angles
| Data: | ISIZE, IORDER, CELEM, RTORS | ||||||||||||||||||||||
| Format: | 2I3, 1X, A4, F9.3 | ||||||||||||||||||||||
| Description: | |||||||||||||||||||||||
| ISIZE | I3 | Number of atoms in ring, or 0 for next ISIZE-1 records | |||||||||||||||||||||
| IORDER | I3 |
|
|||||||||||||||||||||
| CELEM | A4 | Element for atom n | |||||||||||||||||||||
| RTORS | F9.3 | Torsion angle about bond between atoms n and n+1 | |||||||||||||||||||||
Appendix B Pharmacophore File Format
The format used in pharmacophore files has been changed to a comma-separated-variable (CSV) format. This means that pharmacophore data can be loaded directly from the file into a relational database (such as MySQL or ORACLE). In the past, loading the data would have required converting the file written by THINK into a format that the relational database system could handle.
A pharmacophore file begins with two header records (records 1 and 2 below), which are followed by the molecule data blocks. Each block starts with a molecule header record, and then contains the pharmacophore data. Each molecule in the file has a separate data block. All header records start with a "#" character.
In the following record descriptions, the type of each variable is indicated by the initial letter of its name:
| Record 1 (file header) | |||
| Data: | #IREV, IBINS, IMOLS, CCOUNT | ||
| Format: | #I2, I3, I6, 1X, A1, F7.3, 1X, A1 | ||
| Description: | |||
| IREV | I2 | File revision level (currently revision 3) | |
| IBINS | I3 | Number of distance bins | |
| IMOLS | I6 | Number of molecules in file | |
| CCOUNT | A1 | "Y"if data includes pharmacophore counts, otherwise "N" | |
| D3DTOL | F7.3 | 3D tolerance. The pharmacophore tolerance ±x is calculated from 2x = D3DTOL | |
| CNAME | A1 | "Y" if pharmacophore records include molecule names, otherwise "N" | |
| Record 2 (distance bins) | |||
| Data: | #RBIN1, RBIN2, RBIN3, | ||
| Format: | # free format, values separated by spaces | ||
| Description: | |||
| RBINn | Upper limit of distance bin n | ||
| Record 3 (molecule header) | |||
| Data: | #ICENS, IPHARM, ICOUNT, IREC, CMOLE | ||
| Format: | #I3, I11, F11.2, 1X, A | ||
| Description: | |||
| ICENS | I3 | Number of centres in pharmacophores | |
| IPHARM | I11 | Number of unique pharmacophores in molecule | |
| DCOUNT | F11.2 | Total number of pharmacophores for molecule if conformer-based counting is enabled, otherwise 0 | |
| CMOLE | A | Molecule name | |
| Record 4 (pharmacophore data) | |||||||||||||||||||||||||
| Data: | If CNAME=0: CPHARM, RCOUNT If CNAME=1: CMOLE, CPHARM, RCOUNT |
||||||||||||||||||||||||
| Format: | free format, values separated by commas | ||||||||||||||||||||||||
| Description: | |||||||||||||||||||||||||
| CMOLE | Molecule name | ||||||||||||||||||||||||
| CPHARM |
|
||||||||||||||||||||||||
| RCOUNT | Pharmacophore count | ||||||||||||||||||||||||
| D | H-bond donor | R | aromatic ring centroid |
| A | H-bond acceptor | L | lipophile |
| P | positive charge | W | user-defined centre type 1 |
| N | negative charge | X | Lewis base |
| C | acid | M | metal |
| B | base | Z | user-defined centre type 4 |
| d1 | distance between centres x1 and x2 |
| d2 | distance between centres x1 and x3 |
| d3 | distance between centres x1 and x4 |
| d4 | distance between centres x2 and x3 |
| d5 | distance between centres x2 and x4 |
| d6 | distance between centres x3 and x4 |
As a result of the loss of precision which occurs when converting a floating point number into a character string, pharmacophores whose count is less than 0.01 are not written to the file. As a consequence, the sum of the counts in the pharmacophore records may not equal the value of DCOUNT in the molecule header record.
Appendix C Common Variables
The following common variables may be examined (the variable name should be preceded by # to indicate that it is a THINK common variable, rather than a user-defined symbol):
C.1 Counters
| Variable name | Description |
| NTYPES | Number of atom types |
| NMOLES | Number of molecules |
| NATOMS | Number of atoms |
| NFIELD | Number of data fields |
| NTPDUM | Number of dummy atom types |
| NLEARN | Number of records in learn file |
| NFDUSR | Start of user data fields (ie read from files) |
| NTWIST | Number of rotatable bonds |
| NPLTMP | Number of temporary pool addresses in use (internal use) |
| NPHARM | Number of unique pharmacophores in molecule |
| NPDEFN | Number of pharmacophore definitions |
| NRESDS | Number of residues |
| NCYCMX | Maximum number of minimisation cycles allowed during ligand refinement in site searches |
| NITEMX | Maximum number of iterations allowed during ligand refinement in site searches |
C.2 Constants
| Variable name | Description | See section | Default |
| RCWIDT | Current atom label character width | ||
| RCHEIT | Current atom label character height | ||
| RRWIDT | Requested atom label character width | 8.0 | |
| RRHEIT | Requested atom label character height | 12.0 | |
| XLPORG | X-offset to learn plot origin | ||
| YLPORG | Y-offset to learn plot origin | ||
| XLPWID | Learn plot column width | ||
| NPTSNG | Number of conformational points about single bonds | 3 | |
| NPTCON | Number of conformational points about conjugated bonds | 3 | |
| NPTALP | Number of conformational points about alpha bonds | 6 | |
| NPTAMD | Number of conformational points about amide bonds | 0 | |
| NPTRNG | Number of conformational points about ring bonds | 0 | |
| ICSAMP | Conformational analysis sample size | 1000 | |
| IFDACT | Field containing activity data | ||
| SIGACT | Significance value for activity data | 0.3 | |
| IFDXAX | X-axis field for general plot | ||
| IFDYAX | Y-axis field for general plot | ||
| IFDZAX | Z-axis field for general plot | ||
| RATCPK | Ratio of CPK radius to VdW radius | 0.6 | |
| SCUFTR | Display scale factor (pixels per Angstrom) | ||
| NTWUMX | Limit to the number of rotatable bonds for conformer generation (0=no limit) | 0 | |
| RCNFMX | Maximum number of conformers for conformer generation (0=no limit) | 4.1 | 0 |
| D3DTOL | 3D search distance tolerance | 0.5 | |
| D3DVOL | Ratio of pharmacophore volume to number of heavy atoms | 0.25 | |
| D3DARE | Ratio of pharmacophore area to number of heavy atoms | 0.25 | |
| I3DHMX | Limit to number of substructure solutions considered for 3D search | 1000 | |
| ISTHMX | Limit to number of substructure solutions considered for site search | 10000 | |
| I3DSUG | Number of derivatives to be generated for 3D and site searches | 0 | |
| GSTMIN | Maximum acceptable protein-ligand score for conformer to undergo refinement | 8.5 | 1000 |
| GSTHIT | Maximum acceptable score for protein-ligand interactions | 8.5 | 0 |
| IRSEED | Random seed | 90527531 | |
| ICFHMX | Maximum number of hits accepted for each conformer (0=no limit) | 8.4 | 0 |
| IMLHMX | Maximum number of hits accepted for each molecule (0=no limit) | 8.4 | 0 |
| DSINTR | 0 | ||
| TIMEMX | Maximum time for conformational generation during 3D and site searches (minutes) | 60.0 | |
| RADCEN | Overlap between site points and protein atoms (Angstroms) | 9.1 | 0 |
| COZCEN | Cosine of minimum permitted angle for accessibility check | 9.1 | 0.5 |
| RADSIT | Radius of extended active site (Angstroms) | 9.4 | 8.0 |
| GSITE0 | DG0 for conformer score | 8.5 | -5.48 |
| GSTRES | K in contribution to free energy based on fit of conformer to pharmacophore | 8.5 | 1000 |
| GSTHBD | DGhbond for conformer score | 8.5 | -3-34 |
| GSTLIP | DGlipo for conformer score | 8.5 | -0.117 |
| GSTBAD | DGbad for conformer score | 8.5 | 0 |
| ITILSZ | |||
| DMNLOS | Distance used to allocate distance weightings 3 and 4 to site points | 9.5.3 | 6.0 |
| PSCOMN | Minimum acceptable pharmacophore score | 9.5.2 | 10 |
| PSACID | Pharmacophore score penalty for acid-HDON matching | 9.5.4 | 0.3 |
| PSBASE | Pharmacophore score penalty for base-HACC matching | 9.5.4 | 0.5 |
C.3 Flags
| Variable name | Description | See section | Default |
| I3DREP | Bit 0: Skip hydrogens in intramolecular Ligand energy in scoring function | 1 | |
| Bit 1: Skip hydrogen interactions in scoring function | 2 | ||
| Bit 2: Reserved | 0 | ||
| Bit 3: Use softer repulsive term in scoring function | 0 | ||
| Bit 4: Site search Simplex systematic vs random start | 0 | ||
| Bit 5: Site search process only best ICFSMX conformers per molecule | 32 | ||
| Bit 6: Reserved | 0 | ||
| Bit 7: Used by CUSTOM RADIUS=[NO]TOLERANCE | 128 | ||
| IMISNG | Missing value bit pattern | ||
| ICFSMX | Maximum number of best scoring conformers to refine per molecule | 100 | |
| ICOBGD | Background colour for display | 15 | |
| ICOFGD | Foreground colour for display | 16 | |
| ICOACT | Colour for active molecules | 4 | |
| ICOINA | Colour for inactive molecules | 2 | |
| IDGMOL | Update flag for GUI list of molecules | ||
| ICCONT | Conformational analysis contacts check (0=ignore; 1=CPK; 2=VdW) | 1 | |
| ICMODE | Conformational analysis search mode (1=systematic; 2=random; 3=sample) | 1 | |
| IDSMOD | Display mode (internal use) | ||
| ILBMOD | Atom label options (internal use) | ||
| IDVMOD | Diversity display mode (internal use) | ||
| IKYMOD | Key display mode (internal use) | ||
| IMKRNG | Used for conformer generation of ring coordinates (see code) | ||
| IDSCNF | Conformer being displayed | ||
| NDSCNF | Number of conformers being displayed | ||
| KDSMOL | Pool address of list of molecules being displayed (internal use) | ||
| IQUMOL | Query molecule number | ||
| I3DDUM | Bit mask controlling calculation of aromatic ring centroids and lipophiles | 3 | |
| IDSTIL | Current molecule display tile | ||
| NDSTIL | Number of molecule display tiles | ||
| MDXTIL | Number of horizontal tiles in window | ||
| MDYTIL | Number of vertical tiles in window | ||
| IREMOT | Remote property calculation flag | 3.3 | 0 |
| IUDMOD | United Devices mode flag | ||
| NSHITS | Number of hits found in the most recent search | ||
| NSTOTL | Number of molecules searched in the most recent search | ||
| NPHCEN | Number of centres in a pharmacophore | 4 | |
| NPHBIN | Number of pharmacophore distance bins | 10 | |
| IPHCEN | Bit mask of centre types acceptable in pharmacophores | 32748 | |
| IPHMOD | Bit mask controlling pharmacophore calculations | 0 | |
| IHALT | Enables/disables pop-up message box on error | ||
| ISUMOD | De novo compatibility flag (=3 for THINK v1.0) | 0 | |
| ITPACT | Bit mask controlling interpretation of activity values | 0 | |
| ISERVE | Client mode flag (internal use) | ||
| ISTALL | Save best hit only or all hits during site search (0=best; 1=all) | 8.5 | 0 |
| ITRACE | Bit mask controlling production of additional tracing output | D | 0 |
Some of these variables may be altered through the LET command. However, it is recommended that the user checks with Treweren Consultants before attempting to modify any common variable, since alteration of some variables (eg the counters) could have catastrophic consequences.
The common variable ITRACE controls the production of additional output during various THINK operations. This output is often voluminous and is not required during normal usage. However, it can be very helpful when trying to understand exactly why THINK is behaving in a certain manner (eg rejecting molecules during a search).
ITRACE is a bit mask, which allows the controls to be enabled individually. By default it is set to zero (all additional output disabled). To enable output in more than one area, the corresponding values from the table below should be added together, and ITRACE set to the resulting value. For instance, to enable search and profile output, ITRACE should be set to 1025 (1+1024).
| Bit | Value | Purpose | |||||||||||
| 0 | 1 | Search explanations (reasons why molecules are rejected during a search):
|
|||||||||||
| 1 | 2 | Geometry matching (can be very voluminous) | |||||||||||
| 2 | 4 | Graphical monitoring of search refinement | |||||||||||
| 3 | 8 | List atom matching possibilities | |||||||||||
| 4 | 16 | Debugging substructure matching and backtracking during searches (used with bit 10; not recommended for users) | |||||||||||
| 5 | 32 | Reserved for internal use | |||||||||||
| 6 | 64 | Debugging memory pool usage (not recommended for users) | |||||||||||
| 7 | 128 | Debugging trace for error status | |||||||||||
| 8 | 256 | Reserved for internal use | |||||||||||
| 9 | 512 | Reserved for internal use | |||||||||||
| 10 | 1024 | Profile output showing the time spent in various steps (eg during searches) - this also shows which steps were actually executed |
All other bits are reserved for internal use.