Active Volume Constraints
The pharmacophore profiling functionality in THINK uses pharmacophore populations rather than a bit mask. Classifying pharmacophores by distance normally leads to problems when the distance is close to the classification or bin boundary. Pharmacophores with almost identical distances either side of a boundary would be considered different. In THINK this is avoided by distributing the pharmacophore between adjacent bins reflecting the proximity to the bin boundary. The implementation also allows the energy dependent Boltzman populations of the conformations to be used.
For a set of active molecules, the pharmacophores which occur most frequently in a pharmacophore profile can be used as search queries to generate the 3?D coordinates for conformers fitted to that pharmacophore. If each molecule has 4 conformers which exhibit the pharmacophore then for 8 active molecules there are 65536 permutations of conformers, each of which has a corresponding union volume map. In general, it is reasonable to assume that for each pharmacophore the most credible union volume map is the one with the smallest volume corresponding to different molecules occupying the same space.
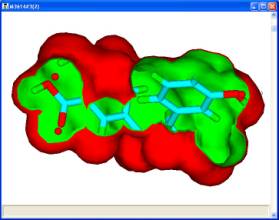
The union volume map can be used as a constraint in a pharmacophore search of a database or library. The use of such constraints eliminates a significant number of false positives during a search. Such maps which represent the volume used by active molecules are known as active volume constraint maps or ligand maps.
Understanding False Positives
At the February SMI Drug Design VI meeting in London, Keith Davies shared an insight into why Structure Based Virtual Screening gives a large proportion of false positives. The results presented suggested that there is a significant difference between the protein crystal conformation (which might be important in molecular recognition) and the protein active conformation. This contrasts with the unproven popular belief that the methods used are too inaccurate.
The paper is also being presented at the Sheffield MGMS/CSAT Conference on Chemoinformatics June 18-9 and the SelectBiosciences Virtual Discovery Meeting in London 24-5 October.
De Novo Derivative Generation
One of the most popular features of the THINK software is its ability to generate de novo derivatives of known molecules. The implementation applies randomly selected transformations to a molecule. The default transformations include oxidations, reductions, additions and substitutions as well as chain elongation and shortening. Many of these correspond to chemical reactions that can be performed which helps to ensure that the molecules are synthetically feasible.
The resulting molecules are automatically filtered by "drug-like", toxicity, metabolism and reactivity criteria which removes most unrealistic molecules. The default filters were suggested by some customers and are similar to those used to select molecules from catalogues for High Throughput Screening.
P450 Project
The range of P450 crystal structures available invites the use of Structure-Based Virtual Screening to predict which molecules might inhibit or be metabolised. However, the P450 family of proteins are known to be flexible and can bind inhibitors away from the metabolic site. Enhancing the existing side-chain flexibility and water functionality of THINK is proving to be an interesting challenge. We would welcome collaborations with scientists with lists of molecules that are inhibitors or substrates.