E. K. Davies
13 July 2005
Most drug molecules mediate their activity by binding to a protein receptor site. When the 3-D structure of the protein is known, then docking or structure-based virtual screening software can be used. However different techniques have to be used, when the crystal structure of the protein has not been determined or the molecules interact with a different conformation of the binding site than occurs in the crystal structure. Molecular fitting techniques to superimpose active molecules and union volumes have been used for many years. For flexible molecules, it can be very time-consuming to manually consider representative conformations of each molecule and find a consistent model for activity. For use in Virtual Screening the superpositioning and the use of volume constraints have to be automated.
THINK uses a concept of a pharmacophore to represent the minimum or essential interaction which an active molecules exhibits for activity. This is formulated in terms of the following atom centered features and the distances between them:
The geometry can be conveniently represented using a "bin model" when a range of distances are stored in the same bin. A 4-centre pharmacophore is then represented by a set of 4 centre types selected from the above list and 6 bin identifiers corresponding to inter-centre distances. The simplest implementation of a distance bin model can be problematic because of bin boundary effects when distances close to a boundary are allocated to one bin but would also occupy a neighbouring bin if the accuracy of the distance was considered. THINK uses a "fuzzy bin model", when the distribution of a distance between bins depends on the proximity to the bin boundary.
The pharmacophores which a molecule exhibits depends on the conformational energy in addition to the features which it contains. THINK can generate a pharmacophore profile for a set of molecules which counts each pharmacophore occurrence. In order to avoid higher energy conformers, the profile applies a Boltzmann population to the pharmacophore counts and the totals are divided by the number of molecules and expressed as a percentage. Thus a pharmacophore which occurs in low energy conformers of all molecules would be expected to have a high pharmacophore profile. Molecules which interact at the same binding site in the same way would be expected to exhibit the same pharmacophore.
For a set of active molecules, the pharmacophores which occur most frequently in a pharmacophore profile can be used as search queries to generate the 3-D coordinates for conformers fitted to that pharmacophore. If there are 2 conformers for each molecule which exhibit the pharmacophore, then for 8 active molecules there are 256 permutations of conformers each of which has a corresponding union volume map. Obviously, for flexible molecules the number of permutations may increase rapidly to computationally prohibitively levels. In general, it is reasonable to assume that for each pharmacophore the most credible union volume map has the smallest volume corresponding to different molecules occupying the same volume. For each pharmacophore, the software considers the molecules in order of increasing number of conformers, retaining the most credible (ie smallest) union volume maps and the corresponding permutation of conformers. In this way, the actually number of permutations is reduced to a manageable number. The best solutions correspond to pharmacophores which are exhibited by the most molecules with small union volume maps.
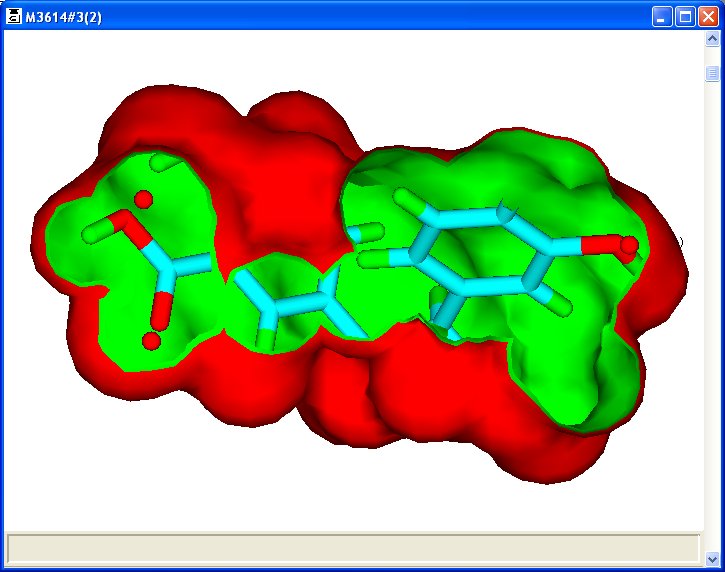
When the union volume map is used with the pharmacophore in a search of a database or library, the map is used as a constraint which represents the volume used by active molecules. Such maps are known as active volume constraint or ligand maps. The use of such constraints eliminates a significant number of false positives during a search. The following (prototype) picture shows an active volume constraint map with pharmacophore centres represented by red circles and an active molecule (clipped).
We currently anticipate this development to be available for beta testing by customers in Q4 of 2005.