Introduction
The cost discovering a new drug and bringing it to market is a major obstacle to addressing and improving human health. Much attention has been focused on the failure rate in clinical trials because of their costs. Over the last 20 years, many pharmaceutical companies have attempted to reduce the number of failures by making decisions earlier about the suitability of molecules as drug candidates. Probably the best example is the Lipinski Rule of 5 [1] which is widely used to avoid synthesis, purchase and testing of molecules which are statistically likely to be problematic during clinical trials due to their physical properties. However, drug failure rates and costs continue to be an obstacle for research companies whilst side-effects and the lack of drug specificity are often a tolerance challenge for patients and their doctors.
The family of P450 enzymes play a crucial role in drug metabolism facilitating their excretion with the inevitable consequence that inhibiting this process can lead to an increased accumulation of drug molecules, as well as molecules produced by the body and those from foods. It is relatively common to read in medication advice leaflets to avoid certain foods or other drugs in order to avoid increased concentrations of a drug which, in the worst case, could have serious toxic side-effects. A review of marketed drugs [2] suggests that the number of inhibitors is comparable to the number of substrates for many P450 isoforms and just over 50% of the inhibitors fail drug-like property tests such as those used by THINK.
Over the last 10 years significant numbers of crystal structures of P450 drug complexes have been solved. Probably the most remarkable aspect of these crystal structures are the changes in side-chain and backbone conformations between very similar proteins which largely account for variety of molecules which bind to these proteins. This poses a major challenge for computational chemistry to predict which molecules bind to the enzyme site and block the metabolic process. It appears that many research organisations [3] respond to this problem during early stage clinical trials by producing derivatives or changing series when significant inhibition is discovered. In some cases, the inhibition is permanent because the drug substrate has facilitated a chemical modification of the protein which can be described as covalent inhibition or suicide inhibitors but is often described by the umbrella term time dependent inhibition reflecting the increasing concentration of inactivated enzymes which are ultimately replaced by newly synthesized proteins.
There is widescale acceptance that the costs and timescale associated with the synthesis and testing of a series of molecules from the initial lead to a clinical candidate have been reduced by predicting whether a molecule has drug-like prior to synthesis or purchase. Obviously, there is scope to extend this philosophy to avoid clinical candidates which interfere with metabolism by binding to P450s. The availability P450 crystal structure data and improved computational techniques prompted Treweren Consultants to investigate whether this was possible with current computer speeds.
Methodology
Over the last 40 years we have observed a dramatic increase in the speed of computers whilst the costs have fallen. However, over the last 10 years the increase in raw computer speeds has been relatively small with more emphasis placed on reduced power consumption and increased memory with 64-bit data architectures. Consequently, we have invested significant effort reviewing the algorithms we developed for docking and scoring in order to improve their performance with some success in part arising from using parallel and multithreaded approaches. Parallel computing is an obvious approach when processing either multiple small molecules or more than one protein binding sites. Our experience with the Oxford University [4] and Find-a-Drug [5] distributing computing projects has demonstrated the power of these approaches. The non-bonding component of the scoring function makes a significant contribution to the optimisation geometry and time taken but is also one of the obvious candidate algorithms for multi-threading.
We created a series of queries from a range of P450 crystal structures on the assumption that any molecule which bound to the iron atom as a Lewis base (or electron donor) as well as at least one other strong interaction (ie at least a 2-centre pharmacophore interaction) might be a potent inhibitor. Most of the originating crystal structures had ligands bound to the heme iron in this way but in a small number of cases we modelled the interaction using standard geometries. To explore this hypothesis, we selected the subset of known inhibitors [2] for the 3A4, 2C9 and 2D6 P450 isoforms. We then docked these molecules into the P450 queries and for a control we used an arbitrary set of 100 catalogue molecules from the distributed computing projects.
Results and Discussion
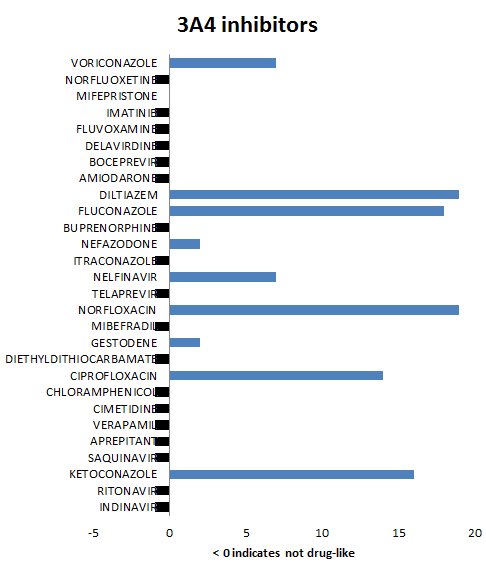
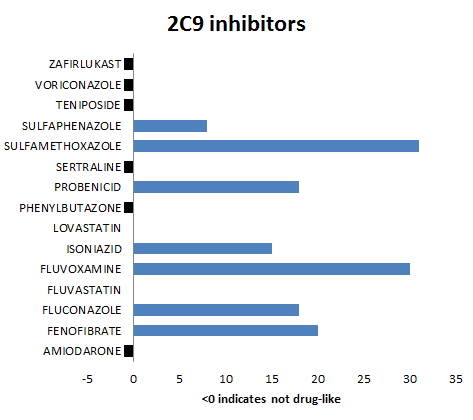
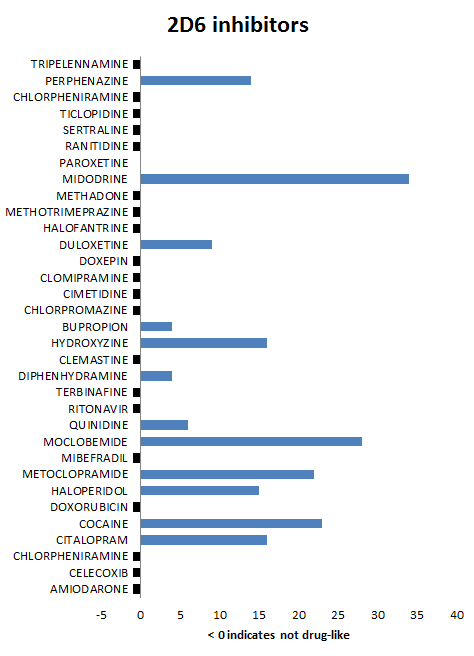
For discussion purposes we present summary results for the 3A4, 2C9 and 2D6 isoforms. The bar charts show the number of P450 queries with which inhibitors are predicted to bind (and for completeness those which fail THINK's standard drug-like test have been allocated a indicator value of -1). The variation in the number of queries to which the inhibitors bind is a reflection of the differences in the queries and is not related to the binding affinity. This also suggests that it is important to use several different crystal structures as the basis of queries for each enzyme in order to span the geometric variation. For the 3A4 and 2D6 isoforms all except one molecule are predicted to bind whilst for the 2C9 isoform the results are not quite as compelling. A review of the queries reveals that none of the 2C9 queries are based on crystal structures where the ligand is bound to the heme iron and it is therefore not surprising that this impacts the quality of the results. Of the 100 control drug-like molecules, 66% were predicted to be inhibitors which is inline with expectations and much lower than the hit rate for known inhibitors. Althought THINK gives a binding score, we have not attempt to correlate this with any estimates of the inhibitor strength partly because of insufficient readily available data but also if one molecule of a series is predicted to be an inhibitor, optimising that series for affinity might also result in increased inhibition: When P450 profiling is being used to reject leads it might be advisible to discard any series where one or more are predicted to be inhibitors.
Conclusion
Predicting P450 inhibition is sufficiently accurate to be used to select leads for optimisation which are significantly less likely to cause drug-drug interactions with the associated potential for toxicity. In the future, additional crystal structures may allow similar methodology to make predictions with greater precision. However, the computer time used is likely to remain longer than that for virtual screening against a single protein site because the potential to dock is evaluated over a set of P450 queries.